Learning Outcomes
- List basic techniques to manipulation genetic information (DNA and RNA)
To understand the basic techniques used to work with nucleic acids, remember that nucleic acids are macromolecules made of nucleotides (a sugar, a phosphate, and a nitrogenous base) linked by phosphodiester bonds. The phosphate groups on these molecules each have a net negative charge. An entire set of DNA molecules in the nucleus is called the genome. DNA has two complementary strands linked by hydrogen bonds between the paired bases. The two strands can be separated by exposure to high temperatures (DNA denaturation) and can be reannealed by cooling. The DNA can be replicated by the DNA polymerase enzyme. Unlike DNA, which is located in the nucleus of eukaryotic cells, RNA molecules leave the nucleus. The most common type of RNA that is analyzed is the messenger RNA (mRNA) because it represents the protein-coding genes that are actively expressed. However, RNA molecules present some other challenges to analysis, as they are often less stable than DNA.
DNA and RNA Extraction
To study or manipulate nucleic acids, the DNA or RNA must first be isolated or extracted from the cells. Various techniques are used to extract different types of DNA (Figure 1). Most nucleic acid extraction techniques involve steps to break open the cell and use enzymatic reactions to destroy all macromolecules that are not desired (such as degradation of unwanted molecules and separation from the DNA sample). Cells are broken using a lysis buffer (a solution which is mostly a detergent); lysis means “to split.” These enzymes break apart lipid molecules in the cell membranes and nuclear membranes. Macromolecules are inactivated using enzymes such as proteases that break down proteins, and ribonucleases (RNAses) that break down RNA. The DNA is then precipitated using alcohol. Human genomic DNA is usually visible as a gelatinous, white mass. The DNA samples can be stored frozen at –80°C for several years.
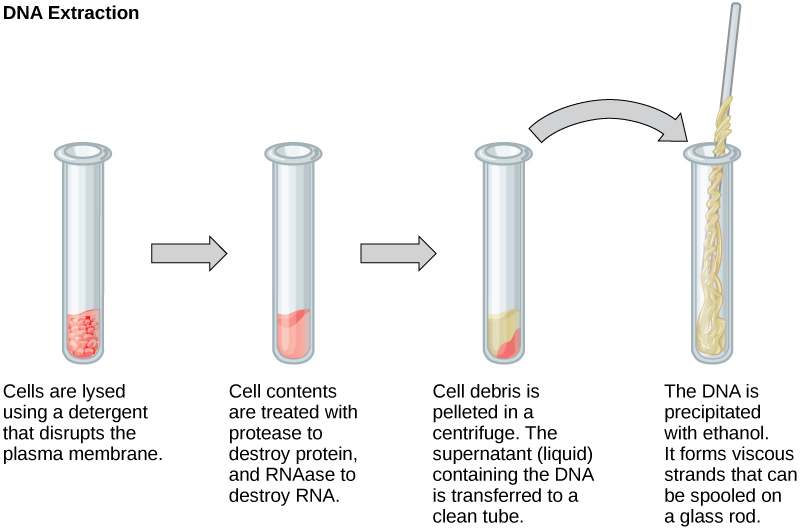
Figure 1. This diagram shows the basic method used for extraction of DNA.
RNA analysis is performed to study gene expression patterns in cells. RNA is naturally very unstable because RNAses are commonly present in nature and very difficult to inactivate. Similar to DNA, RNA extraction involves the use of various buffers and enzymes to inactivate macromolecules and preserve the RNA.

Figure 2. Shown are DNA fragments from seven samples run on a gel, stained with a fluorescent dye, and viewed under UV light. (credit: James Jacob, Tompkins Cortland Community College)
Gel Electrophoresis
Because nucleic acids are negatively charged ions at neutral or basic pH in an aqueous environment, they can be mobilized by an electric field. Gel electrophoresis is a technique used to separate molecules on the basis of size, using this charge. The nucleic acids can be separated as whole chromosomes or fragments. The nucleic acids are loaded into a slot near the negative electrode of a semisolid, porous gel matrix and pushed toward the positive electrode at the opposite end of the gel. Smaller molecules move through the pores in the gel faster than larger molecules; this difference in the rate of migration separates the fragments on the basis of size. There are molecular weight standard samples that can be run alongside the molecules to provide a size comparison. Nucleic acids in a gel matrix can be observed using various fluorescent or colored dyes. Distinct nucleic acid fragments appear as bands at specific distances from the top of the gel (the negative electrode end) on the basis of their size (Figure 2). A mixture of genomic DNA fragments of varying sizes appear as a long smear, whereas uncut genomic DNA is usually too large to run through the gel and forms a single large band at the top of the gel.
Amplification of Nucleic Acid Fragments by Polymerase Chain Reaction
Although genomic DNA is visible to the naked eye when it is extracted in bulk, DNA analysis often requires focusing on one or more specific regions of the genome. Polymerase chain reaction (PCR) is a technique used to amplify specific regions of DNA for further analysis (Figure 3). PCR is used for many purposes in laboratories, such as the cloning of gene fragments to analyze genetic diseases, identification of contaminant foreign DNA in a sample, and the amplification of DNA for sequencing. More practical applications include the determination of paternity and detection of genetic diseases.

Figure 3. Polymerase chain reaction, or PCR, is used to amplify a specific sequence of DNA. Primers—short pieces of DNA complementary to each end of the target sequence—are combined with genomic DNA, Taq polymerase, and deoxynucleotides. Taq polymerase is a DNA polymerase isolated from the thermostable bacterium Thermus aquaticus that is able to withstand the high temperatures used in PCR. Thermus aquaticus grows naturally in the Lower Geyser Basin of Yellowstone National Park. Reverse transcriptase PCR (RT-PCR) is similar to PCR, but cDNA is made from an RNA template before PCR begins.
DNA fragments can also be amplified from an RNA template in a process called reverse transcriptase PCR (RT-PCR). The first step is to recreate the original DNA template strand (called cDNA) by applying DNA nucleotides to the mRNA. This process is called reverse transcription. This requires the presence of an enzyme called reverse transcriptase. After the cDNA is made, regular PCR can be used to amplify it.
Hybridization, Southern Blotting, and Northern Blotting
Nucleic acid samples, such as fragmented genomic DNA and RNA extracts, can be probed for the presence of certain sequences. Short DNA fragments called probes are designed and labeled with radioactive or fluorescent dyes to aid detection. Gel electrophoresis separates the nucleic acid fragments according to their size. The fragments in the gel are then transferred onto a nylon membrane in a procedure called blotting (Figure 4). The nucleic acid fragments that are bound to the surface of the membrane can then be probed with specific radioactively or fluorescently labeled probe sequences. When DNA is transferred to a nylon membrane, the technique is called Southern blotting, and when RNA is transferred to a nylon membrane, it is called northern blotting. Southern blots are used to detect the presence of certain DNA sequences in a given genome, and northern blots are used to detect gene expression. Note that “Southern” blotting is capitalized but no other type of blotting; Southern blotting is named after the scientist who pioneered this technique, Edwin Southern. The other types of blotting were named in reference to the original Southern blot.

Figure 4. Southern blotting is used to find a particular sequence in a sample of DNA. DNA fragments are separated on a gel, transferred to a nylon membrane, and incubated with a DNA probe complementary to the sequence of interest. Northern blotting is similar to Southern blotting, but RNA is run on the gel instead of DNA. In western blotting, proteins are run on a gel and detected using antibodies.
Try It
Candela Citations
- Biology. Provided by: OpenStax CNX. Located at: http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@10.8. License: CC BY: Attribution. License Terms: Download for free at http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@10.8
