Objectives
After completing this section, you should be able to
- describe the bonding and geometry of the carbon-carbon triple bond in terms of the sp-hybridization of the carbon atoms involved.
- explain the reactivity of alkynes based on the known strengths of carbon-carbon single, double and triple bonds.
- write equations for the reaction of an alkyne with one or two equivalents of halogen (chlorine or bromine) or halogen acid (HCl, HBr or HI).
- draw the structure of the product formed when an alkyne reacts with one equivalent of the halogens and halogen acids listed in Objective 3.
- identify the alkyne which must have been used in an addition reaction with a halogen or halogen acid, given the product of such a reaction.
Study Notes
You might find it useful to review Section 1.9 before you begin work on this chapter. If necessary, construct a molecular model of a simple alkyne. Notice the similarity between the behaviour of alkenes and that of alkynes. In the laboratory, you will observe that alkynes readily decolourize a solution of bromine in dichloromethane. Section 9.7 describes a test that allows you to distinguish between a terminal alkyne (i.e., one in which the triple bond occurs between the last two carbons in the chain) and nonterminal alkynes and alkenes.
sup>2-hybridized.
Addition by Electrophilic Reagents
A carbon-carbon triple bond may be located at any unbranched site within a carbon chain or at the end of a chain, in which case it is called terminal. Because of its linear configuration (the bond angle of a sp-hybridized carbon is 180º), a ten-membered carbon ring is the smallest that can accommodate this function without excessive strain. Since the most common chemical transformation of a carbon-carbon double bond is an addition reaction, we might expect the same to be true for carbon-carbon triple bonds. Indeed, most of the alkene addition reactions also take place with alkynes, and with similar regio- and stereoselectivity.
When the addition reactions of electrophilic reagents, such as strong Brønsted acids and halogens, to alkynes are studied we find a curious paradox. The reactions are even more exothermic than the additions to alkenes, and yet the rate of addition to alkynes is slower by a factor of 100 to 1000 than addition to equivalently substituted alkenes. The reaction of one equivalent of bromine with 1-penten-4-yne, for example, gave 4,5-dibromo-1-pentyne as the chief product.
HC≡C-CH2-CH=CH2 + Br2 → HC≡C-CH2-CHBrCH2Br
Although these electrophilic additions to alkynes are sluggish, they do take place and generally display Markovnikov Rule regioselectivity and anti-stereoselectivity. One problem, of course, is that the products of these additions are themselves substituted alkenes and can therefore undergo further addition. Because of their high electronegativity, halogen substituents on a double bond act to reduce its nucleophilicity, and thereby decrease the rate of electrophilic addition reactions. Consequently, there is a delicate balance as to whether the product of an initial addition to an alkyne will suffer further addition to a saturated product. Although the initial alkene products can often be isolated and identified, they are commonly present in mixtures of products and may not be obtained in high yield. The following reactions illustrate many of these features. In the last example, 1,2-diodoethene does not suffer further addition inasmuch as vicinal-diiodoalkanes are relatively unstable.

As a rule, electrophilic addition reactions to alkenes and alkynes proceed by initial formation of a pi-complex, in which the electrophile accepts electrons from and becomes weakly bonded to the multiple bond. Such complexes are formed reversibly and may then reorganize to a reactive intermediate in a slower, rate-determining step. Reactions with alkynes are more sensitive to solvent changes and catalytic influences than are equivalent alkenes. For examples and a discussion of mechanisms click here.
Why are the reactions of alkynes with electrophilic reagents more sluggish than the corresponding reactions of alkenes? After all, addition reactions to alkynes are generally more exothermic than additions to alkenes, and there would seem to be a higher π-electron density about the triple bond ( two π-bonds versus one ). Two factors are significant in explaining this apparent paradox. First, although there are more π-electrons associated with the triple bond, the sp-hybridized carbons exert a strong attraction for these π-electrons, which are consequently bound more tightly to the functional group than are the π-electrons of a double bond. This is seen in the ionization potentials of ethylene and acetylene.
| Acetylene | HC≡CH + Energy → [HC≡CH •(+) + e(–) | ΔH = +264 kcal/mole | |
|---|---|---|---|
| Ethylene | H2C=CH2 + Energy → [H2C=CH2] •(+) + e(–) | ΔH = +244 kcal/mole | |
| Ethane | H3C–CH3 + Energy → [H3C–CH3] •(+) + e(–) | ΔH = +296 kcal/mole |
As defined by the preceding equations, an ionization potential is the minimum energy required to remove an electron from a molecule of a compound. Since pi-electrons are less tightly held than sigma-electrons, we expect the ionization potentials of ethylene and acetylene to be lower than that of ethane, as is the case. Gas-phase proton affinities show the same order, with ethylene being more basic than acetylene, and ethane being less basic than either. Since the initial interaction between an electrophile and an alkene or alkyne is the formation of a pi-complex, in which the electrophile accepts electrons from and becomes weakly bonded to the multiple bond, the relatively slower reactions of alkynes becomes understandable.
A second factor is presumed to be the stability of the carbocation intermediate generated by sigma-bonding of a proton or other electrophile to one of the triple bond carbon atoms. This intermediate has its positive charge localized on an unsaturated carbon, and such vinyl cations are less stable than their saturated analogs. Indeed, we can modify our earlier ordering of carbocation stability to include these vinyl cations in the manner shown below. It is possible that vinyl cations stabilized by conjugation with an aryl substituent are intermediates in HX addition to alkynes of the type Ar-C≡C-R, but such intermediates are not formed in all alkyne addition reactions.
|
Application of the Hammond postulate indicates that the activation energy for the generation of a vinyl cation intermediate would be higher than that for a lower energy intermediate. This is illustrated for alkenes versus alkynes by the following energy diagrams.

Despite these differences, electrophilic additions to alkynes have emerged as exceptionally useful synthetic transforms.
Addition of Hydrogen Halide to an Alkyne
Summary: Reactivity order of hydrogen halides: HI > HB r> HCl > HF.
Follows Markovnikov’s rule:
- Hydrogen adds to the carbon with the greatest number of hydrogens, the halogen adds to the carbon with fewest hydrogens.
- Protination occurs on the more stable carbocation. With the addition of HX, haloalkenes form.
- With the addition of excess HX, you get anti addition forming a geminal dihaloalkane.
Addition of a HX to an Internal Alkyne
As described in Figure 1, the $$\pi$$ electrons will attack the hydrogen of the HBr and because this is a symmetric molecule it does not matter which carbon it adds to, but in an asymmetric molecule the hydrogen will covalently bond to the carbon with the most hydrogens. Once the hydrogen is covalently bonded to one of the carbons, you will get a carbocation intermediate (not shown, but will look the same as depicted in Figure 1) on the other carbon. Again, this is a symmetric molecule and if it were asymmetric, which carbon would have the positive charge?
The final step is the addition of the Bromine, which is a good nucleophile because it has electrons to donate or share. Bromine, therefore attacks the carbocation intermediate placing it on the highly substituted carbon. As a result, you get 2-bromobutene from your 2-butyne reactant, as shown below.
.bmp?revision=1&size=bestfit&width=442&height=160#fixme)
Now, what if you have excess HBr?
Addition due to excess HX yields a geminal dihaloalkane
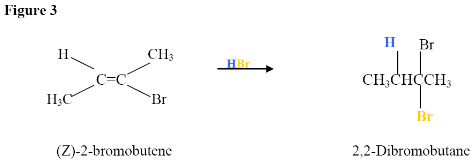
Here, the electrophilic addition proceeds with the same steps used to achieve the product in Addition of a HX to an Internal Alkyne. The $$\pi$$ electrons attacked the hydrogen, adding it to the carbon on the left (shown in blue). Why was hydrogen added to the carbon on left and the one on the right bonded to the Bromine?
Now, you will have your carbocation intermediate, which is followed by the attack of the Bromine to the carbon on the right resulting in a haloalkane product.
Addition of HX to Terminal Alkyne
- Here is an addition of HBr to an asymmetric molecule.
- First, try to make sense of how the reactant went to product and then take a look at the mechanism.

The $$\pi$$ electrons are attacking the hydrogen, depicted by the electron pushing arrows and the Bromine gains a negative charge. The carbocation intermediate forms a positive charge on the left carbon after the hydrogen was added to the carbon with the most hydrogen substituents.
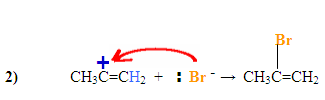
The Bromine, which has a negative charge, attacks the positively charged carbocation forming the final product with the nucleophile on the more substituted carbon.
Addition due to excess HBr present
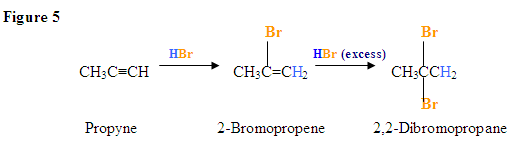
Most Hydrogen halide reactions with alkynes occur in a Markovnikov-manner in which the halide attaches to the most substituted carbon since it is the most positively polarized. A more substituted carbon has more bonds attached to 1) carbons or 2) electron-donating groups such as Fluorine and other halides. However, there are two specific reactions among alkynes where anti-Markovnikov reactions take place: the radical addition of HBr and Hydroboration Oxidation reactions. For alkynes, an anti-Markovnikov addition takes place on a terminal alkyne, an alkyne on the end of a chain.
HBr Addition With Radical Yields 1-bromoalkene
The Br of the Hydrogen Bromide (H-Br) attaches to the less substituted 1-carbon of the terminal alkyne shown below in an anti-Markovnikov manner while the Hydrogen proton attaches to the second carbon. As mentioned above, the first carbon is the less substituted carbon since it has fewer bonds attached to carbons and other substituents. The H-Br reagent must also be reacted with heat or some other radicial initiator such as a peroxide in order for this reaction to proceed in this manner. This presence of the radical or heat leads to the anti-Markovnikov addition since it produces the most stable reaction. For more on Anti-Markovnikov additions:Radical Additions–Anti-Markovnikov Product Formation

The product of a terminal alkyne that is reacted with a peroxide (or light) and H-Br is a 1-bromoalkene.
Regioselectivity: The Bromine can attach in a syn or anti manner which means the resulting alkene can be both cis and trans. Syn addition is when both Hydrogens attach to the same face or side of the double bond (i.e. cis) while the anti addition is when they attach on opposite sides of the bond (trans).

Reaction: Halogenation of Alkynes
Summary:

- Stereoslectivity: anti addition
- Reaction proceeds via cyclic halonium ion
Addition of Br2
- The addition of Br2 to an alkyne is analogous to adding Br2 to an alkene.
- Once Br2 approaches the nucleophilic alkyne, it becomes polarized.
- The $$\pi$$ electrons, from the triple bond, can now attack the polarized bromine forming a C-Br bond and displacing the bromine ion.
- Now, you will get an intermediate electrophilic carbocation, which will immediately react with the bromine ion giving you the dibromo product.
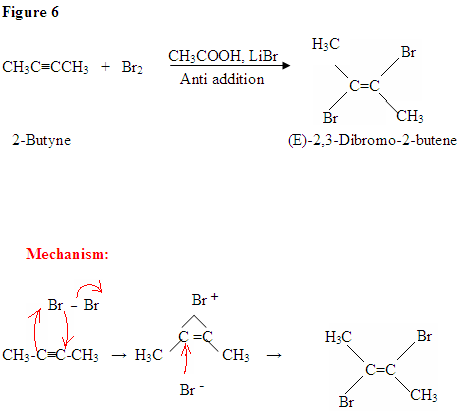
First, you see the polarized Br2 being attacked by the $$\pi$$ electrons. Once you form the C-Br bond, the other bromine is released as a bromine ion. The intermediate here is a bromonium ion, which is electrophilic and reacts with the bromine ion giving you the dibromo product.
Exercise
Draw the structure, and give the IUPAC name, of the product formed in each of the reactions listed below.
- $\ce{\sf{CH3-C#C-CH3->[\displaystyle{\textrm{1 equiv}}][\displaystyle{\textrm{HCl}}]}}$
- $\ce{\sf{CH3-C#C-CH3->[\displaystyle{\textrm{excess}}][\displaystyle{\textrm{HCl}}]}}$
- $\ce{\sf{CH3-C#C-CH3->[\displaystyle{\textrm{1 equiv}}][\displaystyle{\textrm{Br}_2}]}}$
- $\ce{\sf{CH3-C#C-CH3->[\displaystyle{\textrm{excess}}][\displaystyle{\textrm{Br}_2}]}}$
- $\ce{\sf{CH3CH2-C#C-H->[\displaystyle{\textrm{1 equiv}}][\displaystyle{\textrm{HCl}}]}}$
- $\ce{\sf{CH3CH2-C#C-H->[\displaystyle{\textrm{excess}}][\displaystyle{\textrm{HCl}}]}}$
- $\ce{\sf{CH3CH2CH2-C#C-H->[\displaystyle{\textrm{1 equiv}}][\displaystyle{\textrm{Br}_2}]}}$
- $\ce{\sf{CH3CH2CH2-C#C-H->[\displaystyle{\textrm{excess}}][\displaystyle{\textrm{Br}_2}]}}$
Answer
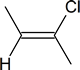
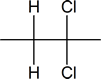
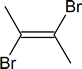
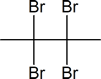



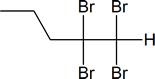
Contributors
- Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
- Prof. Steven Farmer (Sonoma State University)
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
- Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)
- Jim Clark (Chemguide.co.uk)