The three phases of radical chain reactions
The following video by Leah4Sci provides a good introduction to free radical reactions.
https://www.youtube.com/watch?v=Uz1_n9ZnksY
Because of their high reactivity, free radicals have the potential to be both extremely powerful chemical tools and extremely harmful contaminants. Much of the power of free radical species stems from the natural tendency of radical processes to occur in a chain reaction fashion. Radical chain reactions have three distinct phases: initiation, propagation, and termination.

The initiation phase describes the step that initially creates a radical species. In most cases, this is a homolytic cleavage event, and takes place very rarely due to the high energy barriers involved. Often the influence of heat, UV radiation, or a metal-containing catalyst is necessary to overcome the energy barrier.
Molecular chlorine and bromine will both undergo homolytic cleavage to form radicals when subjected to heat or light. Other functional groups which also tend to form radicals when exposed to heat or light are chlorofluorocarbons, peroxides, and the halogenated amide N-bromosuccinimide (NBS).

The propagation phase describes the ‘chain’ part of chain reactions. Once a reactive free radical is generated, it can react with stable molecules to form new free radicals. These new free radicals go on to generate yet more free radicals, and so on. Propagation steps often involve hydrogen abstraction or addition of the radical to double bonds.

Chain termination occurs when two free radical species react with each other to form a stable, non-radical adduct. Although this is a very thermodynamically downhill event, it is also very rare due to the low concentration of radical species and the small likelihood of two radicals colliding with one another. In other words, the Gibbs free energy barrier is very high for this reaction, mostly due to entropic rather than enthalpic considerations. The active sites of enzymes, of course, can evolve to overcome this entropic barrier by positioning two radical intermediates adjacent to one another.
Radical halogenation in the lab
The chlorination of an alkane provides a simple example of a free radical chain reaction. In the initiation phase, a chlorine molecule undergoes homolytic cleavage after absorbing energy from light:
![]()
The chlorine radical then abstracts a hydrogen, leading to an alkyl radical (step 2), which reacts with a second chlorine molecule (step 3) to form the chloroalkane product plus chlorine radical, which then returns to repeat step 2.


Likely chain termination steps are the condensation of two alkyl radical intermediates or condensation of an alkane radical with a chlorine radical.
Alkane halogenation reactions exhibit a degree of regiospecificity: if 2-methylbutane is subjected to a limiting amount of chlorine, for example, chlorination takes place fastest at the tertiary carbon.

This is because the tertiary radical intermediate is more stable than the secondary radical intermediate that results from abstraction of the proton on carbon #3, and of course both are more stable than a primary radical intermediate. Recall that the Hammond postulate (section 6.2, section 15.2B) tells us that a lower-energy intermediate implies a lower-energy transition state, and thus a faster reaction.
Unfortunately, chloroalkanes will readily undergo further chlorination resulting in polychlorinated products, so this is not generally a terribly useful reaction from a synthetic standpoint.
Alkanes can be brominated by a similar reaction. The regiochemical trends are the same as for chlorination, but significantly more pronounced (in other words, bromination is more regioselective). This is because hydrogen abstraction by bromine radical is much less exergonic than by chorine radical – and this in turn means that the transition state for abstraction by bromine resembles the resulting intermediate more closely than the transition state for abstraction by chlorine resembles its intermediate.

Another way of saying the same thing is that the bromination transition state has more ‘radical character’ than the chlorination transition state. Trends in radical stability thus have a greater influence on the speed of hydrogen abstraction.
The reaction proceeds through the radical chain mechanism. The radical chain mechanism is characterized by three steps: initiation, propagation and termination. Initiation requires an input of energy but after that the reaction is self-sustaining. The first propagation step uses up one of the products from initiation, and the second propagation step makes another one, thus the cycle can continue until indefinitely.
Step 1: Initiation
Initiation breaks the bond between the chlorine molecule (Cl2). For this step to occur energy must be put in, this step is not energetically favorable. After this step, the reaction can occur continuously (as long as reactants provide) without input of more energy. It is important to note that this part of the mechanism cannot occur without some external energy input, through light or heat.

Step 2: Propagation
The next two steps in the mechanism are called propagation steps. In the first propagation step, a chlorine radical combines with a hydrogen on the methane. This gives hydrochloric acid (HCl, the inorganic product of this reaction) and the methyl radical. In the second propagation step more of the chlorine starting material (Cl2) is used, one of the chlorine atoms becomes a radical and the other combines with the methyl radical.

The first propagation step is endothermic, meaning it takes in heat (requires 2 kcal/mol) and is not energetically favorable. In contrast the second propagation step is exothermic, releasing 27 kcal/mol. Since the second propagation step is so exothermic, it occurs very quickly. The second propagation step uses up a product from the first propagation step (the methyl radical) and following Le Chatelier’s principle, when the product of the first step is removed the equilibrium is shifted towards it’s products. This principle is what governs the unfavorable first propagation step’s occurance.

Step 3: Termination
In the termination steps, all the remaining radicals combine (in all possible manners) to form more product (CH3Cl), more reactant (Cl2) and even combinations of the two methyl radicals to form a side product of ethane (CH3CH3).

Problems with the chlorination of methane
The chlorination of methane does not necessarily stop after one chlorination. It may actually be very hard to get a monosubstituted chloromethane. Instead di-, tri- and even tetra-chloromethanes are formed. One way to avoid this problem is to use a much higher concentration of methane in comparison to chloride. This reduces the chance of a chlorine radical running into a chloromethane and starting the mechanism over again to form a dichloromethane. Through this method of controlling product ratios one is able to have a relative amount of control over the product.
Chlorination of other alkanes
When alkanes larger than ethane are halogenated, isomeric products are formed. Thus chlorination of propane gives both 1-chloropropane and 2-chloropropane as mono-chlorinated products. Four constitutionally isomeric dichlorinated products are possible, and five constitutional isomers exist for the trichlorinated propanes. Can you write structural formulas for the four dichlorinated isomers?
\[CH_3CH_2CH_3 + 2Cl_2 \rightarrow \text{Four} \; C_3H_6Cl_2 \; \text{isomers} + 2 HCl\]
The halogenation of propane discloses an interesting feature of these reactions. All the hydrogens in a complex alkane do not exhibit equal reactivity. For example, propane has eight hydrogens, six of them being structurally equivalent primary, and the other two being secondary. If all these hydrogen atoms were equally reactive, halogenation should give a 3:1 ratio of 1-halopropane to 2-halopropane mono-halogenated products, reflecting the primary/secondary numbers. This is not what we observe. Light-induced gas phase chlorination at 25 ºC gives 45% 1-chloropropane and 55% 2-chloropropane.
CH3-CH2-CH3 + Cl2 → 45% CH3-CH2-CH2Cl + 55% CH3-CHCl-CH3
The results of bromination ( light-induced at 25 ºC ) are even more surprising, with 2-bromopropane accounting for 97% of the mono-bromo product.
CH3-CH2-CH3 + Br2 → 3% CH3-CH2-CH2Br + 97% CH3-CHBr-CH3
These results suggest strongly that 2º-hydrogens are inherently more reactive than 1º-hydrogens, by a factor of about 3:1. Further experiments showed that 3º-hydrogens are even more reactive toward halogen atoms. Thus, light-induced chlorination of 2-methylpropane gave predominantly (65%) 2-chloro-2-methylpropane, the substitution product of the sole 3º-hydrogen, despite the presence of nine 1º-hydrogens in the molecule.
(CH3)3CH + Cl2 → 65% (CH3)3CCl + 35% (CH3)2CHCH2Cl
If you are uncertain about the terms primary (1º), secondary (2º) & tertiary (3º) Click Here.
It should be clear from a review of the two steps that make up the free radical chain reaction for halogenation that the first step (hydrogen abstraction) is the product determining step. Once a carbon radical is formed, subsequent bonding to a halogen atom (in the second step) can only occur at the radical site. Consequently, an understanding of the preference for substitution at 2º and 3º-carbon atoms must come from an analysis of this first step.
Radical Allylic Halogenation
Watch a video by Organic Chemistry Tutor on allylic halogenation:
https://youtu.be/EUp2c5aO4dk

Why substitution of allylic hydrogens?
As the table below shows, the dissociation energy for the allylic C-H bond is lower than the dissociation energies for the C-H bonds at the vinylic and alkylic positions. This is because the radical formed when the allylic hydrogen is removed is resonance-stabilized. Hence, given that the halogen concentration is low, substitution at the allylic position is favored over competing reactions. However, when the halogen concentration is high, addition at the double bond is favored because a polar reaction outcompetes the radical chain reaction.

Radical Allylic Bromination (Wohl-Ziegler Reaction)
Preparation of Bromine (low concentration)
NBS (N-bromosuccinimide) is the most commonly used reagent to produce low concentrations of bromine. When suspended in tetrachloride (CCl4), NBS reacts with trace amounts of HBr to produce a low enough concentration of bromine to facilitate the allylic bromination reaction.

Allylic Bromination Mechanism
Step 1: Initiation
Once the pre-initiation step involving NBS produces small quantities of Br2, the bromine molecules are homolytically cleaved by light to produce bromine radicals.

Step 2: Propagation
One bromine radical produced by homolytic cleavage in the initiation step removes an allylic hydrogen of the alkene molecule. A radical intermediate is generated, which is stabilized by resonance. The stability provided by delocalization of the radical in the alkene intermediate is the reason that substitution at the allylic position is favored over competing reactions such as addition at the double bond.

The intermediate radical then reacts with a Br2 molecule to generate the allylic bromide product and regenerate the bromine radical, which continues the radical chain mechanism. If the alkene reactant is asymmetric, two distinct product isomers are formed.

Step 3: Termination
The radical chain mechanism of allylic bromination can be terminated by any of the possible steps shown below.

Radical Allylic Chlorination
Like bromination, chlorination at the allylic position of an alkene is achieved when low concentrations of Cl2 are present. The reaction is run at high temperatures to achieve the desired results.
Industrial Uses
Allylic chlorination has important practical applications in industry. Since chlorine is inexpensive, allylic chlorinations of alkenes have been used in the industrial production of valuable products. For example, 3-chloropropene, which is necessary for the synthesis of products such as epoxy resin, is acquired through radical allylic chlorination (shown below).
![]()
Useful polymers formed by radical chain reactions
Many household polymeric materials with which you are probably familiar are made with a radical chain reaction process. Polyethylene (PET), the plastic material used to make soft drink bottles and many other kinds of packaging, is produced by the radical polymerization of ethylene (ethene in IUPAC nomenclature). A radical initiator such as benzoyl peroxide undergoes homolytic cleavage when subjected to high temperatures.

In the propagation phase, the benzoyl radical (X• in the figure below) adds to the double bond of ethylene, generating a new organic radical. Successive ethylene molecules add to the growing polymer, until termination occurs when two radicals happen to collide. In the figure below, the growing PET polymer is terminated by a benzoyl radical, but in an alternative termination step two growing PET radicals could condense.

Other small substituted alkene monomers polymerize in a similar fashion to form familiar polymer materials. Two examples are given below.

Destruction of the ozone layer by CFC radicals
The high reactivity of free radicals and the multiplicative nature of radical chain reactions can be useful in the synthesis of materials such as polyethylene plastic – but these same factors can also result in dangerous consequences. You are probably aware of the danger posed to the earth’s protective stratospheric ozone layer by the use of chlorofluorocarbons (CFCs) as refrigerants and propellants in aerosol spray cans. Freon-11, or CFCl3, is a typical CFC that was widely used until fairly recently. It can take months or years for a CFC molecule to drift up into the stratosphere from the surface of the earth, and of course the concentration of CFCs at this altitude is very low. Ozone, on the other hand, is continually being formed in the stratosphere. Why all the concern, then, about destruction of the ozone layer – how could such a small amount of CFCs possibly do significant damage? The problem lies in the fact that the process by which ozone is destroyed is a chain reaction, so that a single CFC molecule can initiate the destruction of many ozone molecules before a chain termination event occurs.
Harmful radical species in cells and natural antioxidants
While the high reactivity of the hydroxide radical is a beneficial trait in the atmosphere, it is a harmful trait when the same hydroxide radical is present in a living cell. Hydroxide radical and other reactive oxygen species (ROS) such as superoxide (O2–) and peroxide (O2 2-) are continuously produced as minor side-products in the reduction of O2 to H2O in respiration.

The ROS are highly reactive oxidizing agents, capable of inflicting damage to DNA, proteins, and the lipids of cell membranes – they are thought to play a major role in the natural aging process. Hydroxide radical, for example, will initiate a radical chain reaction with the hydrocarbon part of an unsaturated membrane lipid molecule that results in the formation of lipid hydroperoxide.
One important antioxidant that you are no doubt familiar with is ascorbic acid, or vitamin C. It reacts with harmful radicals to produce the ascorbyl radical, which is significantly more stable than most other radical species due to resonance delocalization. The end result of this first step is that a very reactive, potentially harmful radical (X•) has been ‘quenched’, and replaced by a much less reactive (and thus less harmful) ascorbyl radical.is thus potentially able to scavenge two harmful radical species.
Dehydroascorbate is subsequently either broken down and excreted, or else recycled (reduced) back to ascorbate. This can happen either in a direct, enzyme-free reaction with glutathione, or through the action of a specific glutathione/NADH-dependant reductase enzyme. You were invited to propose a likely mechanism for the enzyme-free reaction in problem 16.14.
Contributors
- Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)
Further Reading
MasterOrganicChemistry
Radical Reactions – Why Is “Heat” Or “Light” Required?
Initiation, Propagation, Termination
Carey 4th Edition On-Line Activity
Web Pages
Really nice summary of radical reactions
Mechanism of free radical halogenation
Videos
Another radical mechanism video
Steps of a free radical mechanism
Free radical chain mechanism video
Free radical halogenation video
Free radical halogenation video
Free radical halogenation video
Intro video on free radical halogenation
Tutorial
How to draw free radical chain mechanisms
Free radical reaction tutorial
Exercises
QuestionsQ10.2.1
Predict the mono-substituted halogenated product(s) of chlorine gas reacting with 2-methylbutane.
Q10.2.2
Predict the relative amount of each mono-brominated product when 3-methylpentane is reacted with Br2. Consider 1°, 2°, 3° hydrogen.
Solutions
S10.2.1

S10.2.2

Contributors
- Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
- Prof. Steven Farmer (Sonoma State University)
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
- Jim Clark (Chemguide.co.uk)
- Kristen Kelley and Britt Farquharson
References
- Djerassi, Carl. “Brominations with N-Bromosuccinimide and Related Compounds – The Wohl-Ziegler Reaction.” Chemical Reviews 43 (1948):271-314.
- Easton, Christopher J., Alison J. Edwards, Stephen B. McNabb, Martin C. Merrett, Jenny L. O’Connell, Gregory W. Simpson, Jamie S. Simpson, and Anthony C. Willis. “Allylic halogenation of unsaturated amino acids.” Organic and Biomolecular Chemistry (2003). RSC Publishing. 9 June 2003. Royal Society of Chemistry. 25 Feb. 2009.
- Kent, Doug. Allylic Bromination. Chem 118B Workshop. Learning Skills Center. 3 Feb. 2009.
- Li, Chao-Jun, and Tak-Hang Chan. Comprehensive Organic Reactions in Aqueous Media. New York: Wiley-Interscience, 2007.
- Vollhardt, Peter C., and Neil E. Schore. Organic Chemistry: Structure and Function. 5th ed. New York: W.H. Freeman and Company, 2007.
Outside Links
- http://en.wikipedia.org/wiki/N-Bromosuccinimide#Preparation
- http://en.wikipedia.org/wiki/Wohl-Ziegler_reaction
- http://www.mhhe.com/physsci/chemistry/carey/student/olc/graphics/carey04oc/ref/ch10allylic.html
Hydrogen bromide and alkenes: The peroxide effect
A symmetrical alkene is one like ethene where the groups at both ends of the carbon-carbon double bond are the same. The reaction happens at room temperature in the presence of organic peroxides or some oxygen from the air. Alkenes react very slowly with oxygen to produce traces of organic peroxides – so the two possible conditions are equivalent to each other.
The reaction is a simple addition of the hydrogen bromide. For example, with ethene:
![]()
![]()
With a symmetrical alkene you get exactly the same product in the absence of the organic peroxides or oxygen – but the mechanism is different.
The mechanism
Hydrogen halides (hydrogen chloride, hydrogen bromide and the rest) usually react with alkenes using an electrophilic addition mechanism. However, in the presence of organic peroxides, hydrogen bromide adds by a different mechanism.
| Note |
|---|
| If you are interested, you will find the electrophilic addition mechanism for the addition of hydrogen bromide and other hydrogen halides to alkenes if you follow this link. You may need to explore several pages in this section. |
With the organic peroxides present you get a free radical chain reaction.
Chain initiation
The chain is initiated by free radicals produced by an oxygen-oxygen bond in the organic peroxide breaking.
![]()
![]()
These free radicals extract a hydrogen atom from a hydrogen bromide molecule to produce bromine radicals.
![]()
![]()
Chain propagation
A bromine radical joins to the ethene using one of the electrons in the pi bond. That creates a new radical with the single electron on the other carbon atom.
![]()
![]()
That radical reacts with another HBr molecule to produce bromoethane and another bromine radical to continue the process.
![]()
![]()
![]() etc
etc
Chain termination
Eventually two free radicals hit each other and produce a molecule of some sort. The process stops here because no new free radicals are formed.
Addition to unsymmetrical alkenes
An unsymmetrical alkene is one like propene where the groups at either end of the carbon-carbon double bond are different.The reaction happens under the same conditions as with a symmetrical alkene, but there is a complication because the hydrogen and the bromine can add in two different ways. Which way they add depends on whether there are organic peroxides (or oxygen) present or not.

Normally, when a molecule HX adds to a carbon-carbon double bond, the hydrogen becomes attached to the carbon with the more hydrogens on already. This is known as Markovnikov’s Rule.
Because the HBr adds on the “wrong way around ” in the presence of organic peroxides, this is often known as the peroxide effect or anti-Markovnikov addition.
In the absence of peroxides, hydrogen bromide adds to propene via an electrophilic addition mechanism. That gives the product predicted by Markovnikov’s Rule.
The free radical mechanism
Chain initiation
This is exactly the same as in the ethene case above.
![]()
![]()
![]()
![]()
Chain propagation
When the bromine radical joins to the propene, it attaches so that a secondary radical is formed. This is more stable (and so easier to form) than the primary radical which would be formed if it attached to the other carbon atom.
![]()
![]()
That radical reacts with another HBr molecule to produce 1-bromopropane and another bromine radical to continue the process.
![]()
![]()
![]() etc
etc
Chain termination
Eventually two free radicals hit each other and produce a molecule of some sort. The process stops here because no new free radicals are formed.
Why don’t the other hydrogen halides behave in the same way?
The reason that hydrogen bromide adds in an anti-Markovnikov fashion in the presence of organic peroxides is simply a question of reaction rates. The free radical mechanism is much faster than the alternative electrophilic addition mechanism. Both mechanisms happen, but most of the product is the one from the free radical mechanism because that is working faster. With the other hydrogen halides, the opposite is true.
- Hydrogen fluoride: The hydrogen-fluorine bond is so strong that fluorine radicals aren’t formed in the initiation step.
- Hydrogen chloride: With hydrogen chloride, the second half of the propagation stage is very slow. If you do a bond enthalpy sum, you will find that the following reaction is endothermic.
![]()
![]()
This is due to the relatively high hydrogen-chlorine bond strength.
- Hydrogen iodide: In this case, the first step of the propagation stage turns out to be endothermic and this slows the reaction down. Not enough energy is released when the weak carbon-iodine bond is formed.
![]()
![]()
In the case of hydrogen bromide, both steps of the propagation stage are exothermic.
Contributors
Jim Clark (Chemguide.co.uk)
References
- K. Peter C. Vollhardt, Neil E. Schore; Organic Chemistry: Structure and Function Fifth Edition; W. H. Freeman and Campany, 2007
- Micheal Vokin; Nuffield Advance Chemistry Student’s Book Forth Edition; Person Education Limited, 2004
Outside Links
Problems
Please give the product(s) of the reactions below:
- CH3-C(CH3)=CH-CH3 + HBr + H2O2 ==> ?
- CH3-C(CH3)=CH-CH3 + HI + H2O2 ==> ?
- CH3-C(CH3)=CH-CH3 + HCl + H2O2 ==> ?
- CH3-CH=CH-CH3 + HBr + H2O2 ==> ?
- CH3-C(CH3)=CH-CH3 + HBr ==> ?
Answers
- CH3-CH(CH3)-CHBr-CH3 (Anti-Markovnikov)
- CH3-C(CH3)I-CH2-CH3 (Markovnikov)
- CH3-C(CH3)Cl-CH2-CH3 (Markovnikov)
- CH3-CHBr-CH-CH3 or CH3-CH-CHBr-CH3 (Both molecules are the same)
- CH3-C(CH3)Br-CH2-CH3 (Markovnikov)
References
Contributors
- Kelvin Kan (UCD)
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Reduction of alkynes to trans-alkenes via radical reaction
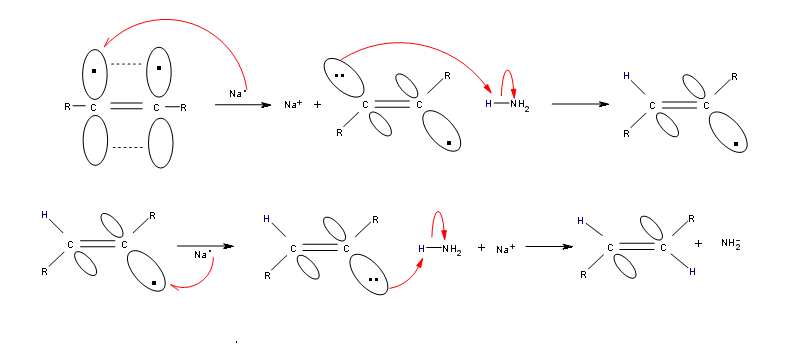
Alkynes can be reduced to trans-alkenes with the use of sodium dissolved in an ammonia solvent. An Na radical donates an electron to one of the P bonds in a carbon-carbon triple bond. This forms an anion, which can be protonated by a hydrogen in an ammonia solvent. This prompts another Na radical to donate an electron to the second P orbital. Soon after this anion is also protonated by a hydrogen from the ammonia solvent, resulting in a trans-alkene.
R-C≡C-R + 2 Na in NH3 (liq) ——> trans R-CH=CH-R + 2 NaNH2
Mechanism
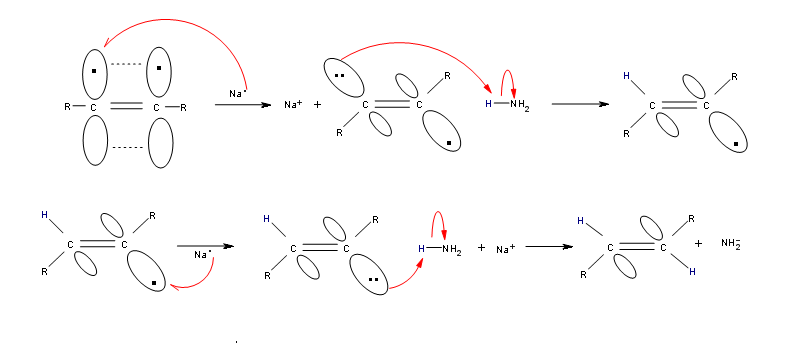 Contributors
Contributors
- Ravjot Takhar (UCD)
- Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
- Prof. Steven Farmer (Sonoma State University)
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Birch reduction
The Birch reduction is an organic reaction which is particularly useful in synthetic organic chemistry. The reaction was reported in 1944 by the Australian chemist Arthur Birch (1915–1995) working in the Dyson Perrins Laboratory at the University of Oxford, building on earlier work by Wooster and Godfrey published in 1937. It converts aromatic compounds having a benzenoid ring into a product, 1,4-cyclohexadienes, in which two hydrogen atoms have been attached on opposite ends of the molecule. It is the organic reduction of aromatic rings in liquid ammonia with sodium, lithium or potassium and an alcohol, such as ethanol and tert-butanol. This reaction is quite unlike catalytic hydrogenation, which usually reduces the aromatic ring all the way to a cyclohexane.
The original reaction reported by Arthur Birch in 1944 used sodium and ethanol.[1][2][3] Alfred L. Wilds later discovered that lithium gives better yields.[8] Also the use of tert-butyl alcohol has become common. The reaction is widely used in synthetic organic chemistry.
An example is the reduction of naphthalene:[9]
Basic reaction mechanism
A solution of sodium in liquid ammonia consists of the electride salt [Na(NH3)x]+ e−, which has an intense blue color. The solvated electrons add to the aromatic ring to give a radical anion. The added alcohol supplies a proton to the radical anion and also to the penultimate carbanion; for most substrates ammonia is not acidic enough.[14]
Candela Citations
- 5.5.tThe Free-Radical Chain Reaction. Authored by: Tim Soderberg. Provided by: University of Minnesota, Morris. Located at: https://chem.libretexts.org/LibreTexts/Purdue/Purdue_Chem_26100%3A_Organic_Chemistry_I_(Wenthold)/Chapter_05%3A_The_Study_of_Chemical_Reactions/5.5.%09The_Free-Radical_Chain_Reaction. Project: Chemistry LibreTexts . License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 6.3 Radical Reactions. Authored by: Dr. Dietmar Kennepohl FCIC; Prof. Steven Farmer; Tim Soderberg. Provided by: Athabasca University; Sonoma State University; University of Minnesota, Morris. Located at: https://chem.libretexts.org/LibreTexts/Athabasca_University/Chemistry_350%3A_Organic_Chemistry_I/Chapter_6%3A_An_Overview_of_Organic_Reactions/6.03_Radical_Reactions. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 10.2: Preparing Alkyl Halides from Alkanes - Radical Halogenation. Authored by: Dr. Dietmar Kennepohl FCIC; Prof. Steven Farmer; William Reusch, Professor Emeritus; Jim Clark; Kristen Kelley and Britt Farquharson. Provided by: Athabasca University; Sonoma State University; Michigan State U. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Map%3A_Organic_Chemistry_(McMurry)/Chapter_10%3A_Organohalides/10.02_Preparing_Alkyl_Halides_from__Alkanes%3A_Radical_Halogenation. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- Radical Allylic Halogenation. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Alkenes/Reactivity_of_Alkenes/Free_Radical_Reactions_of_Alkenes/Radical_Allylic_Halogenation. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- Hydrogen Bromide and Aklenes: The Peroxide Effect. Authored by: Jim Clark. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Polymers/Hydrogen_Bromide_and_Aklenes%3A_The_Peroxide_Effect. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 9:8tRadical Additions: Anti-Markovnikov Product Formation. Authored by: Kevin Kan; William Reusch, Professor Emeritus. Provided by: UCD; Michigan State U. Located at: https://chem.libretexts.org/LibreTexts/Winona_State_University/Klein_and_Straumanis_Guided/9%3A_Addition_Reactions_of_Alkenes/9%3A8%09Radical_Additions%3A_Anti-Markovnikov_Product_Formation. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 9.5: Reduction of Alkynes. Authored by: Ravjot Takhar; Dr. Dietmar Kennepohl FCIC; Prof. Steven Farmer; William Reusch, Professor Emeritus. Provided by: UCD; Athabasca University; Sonoma State University; Michigan State U. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Map%3A_Organic_Chemistry_(McMurry)/Chapter_09%3A_Alkynes%3A_An_Introduction_to_Organic_Synthesis/9.05_Reduction_of_Alkynes. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- Birch Reduction. Authored by: Wikipedia. Provided by: Wikipedia. Located at: https://en.wikipedia.org/wiki/Birch_reduction. Project: Wikipedia. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
