List key technologies enabling modern uses of biology
Biotechnology is the use of biological agents for technological advancement. Biotechnology was used for breeding livestock and crops long before the scientific basis of these techniques was understood. Since the discovery of the structure of DNA in 1953, the field of biotechnology has grown rapidly through both academic research and private companies. The primary applications of this technology are in medicine (production of vaccines and antibiotics) and agriculture (genetic modification of crops, such as to increase nutrient content). Biotechnology also has many industrial applications, such as fermentation, the treatment of oil spills, and the production of biofuels (Figure 1).

Figure 1. Antibiotics are chemicals produced by fungi, bacteria, and other organisms that have antimicrobial properties. The first antibiotic discovered was penicillin. Antibiotics are now commercially produced and tested for their potential to inhibit bacterial growth. (credit “advertisement”: modification of work by NIH; credit “test plate”: modification of work by Don Stalons/CDC; scale-bar data from Matt Russell)
In this outcome, we will learn about some modern technologies used in biology today.
Learning Objectives
- List basic techniques to manipulation genetic information (DNA and RNA)
- Recognize technologies used for molecular, cellular, and reproductive cloning
- Understand the basics of genetic engineering
Manipulating Genetic Material
To understand the basic techniques used to work with nucleic acids, remember that nucleic acids are macromolecules made of nucleotides (a sugar, a phosphate, and a nitrogenous base) linked by phosphodiester bonds. The phosphate groups on these molecules each have a net negative charge. An entire set of DNA molecules in the nucleus is called the genome. DNA has two complementary strands linked by hydrogen bonds between the paired bases. The two strands can be separated by exposure to high temperatures (DNA denaturation) and can be reannealed by cooling. The DNA can be replicated by the DNA polymerase enzyme. Unlike DNA, which is located in the nucleus of eukaryotic cells, RNA molecules leave the nucleus. The most common type of RNA that is analyzed is the messenger RNA (mRNA) because it represents the protein-coding genes that are actively expressed. However, RNA molecules present some other challenges to analysis, as they are often less stable than DNA.
DNA and RNA Extraction
To study or manipulate nucleic acids, the DNA or RNA must first be isolated or extracted from the cells. Various techniques are used to extract different types of DNA (Figure 2). Most nucleic acid extraction techniques involve steps to break open the cell and use enzymatic reactions to destroy all macromolecules that are not desired (such as degradation of unwanted molecules and separation from the DNA sample). Cells are broken using a lysis buffer (a solution which is mostly a detergent); lysis means “to split.” These enzymes break apart lipid molecules in the cell membranes and nuclear membranes. Macromolecules are inactivated using enzymes such as proteases that break down proteins, and ribonucleases (RNAses) that break down RNA. The DNA is then precipitated using alcohol. Human genomic DNA is usually visible as a gelatinous, white mass. The DNA samples can be stored frozen at –80°C for several years.
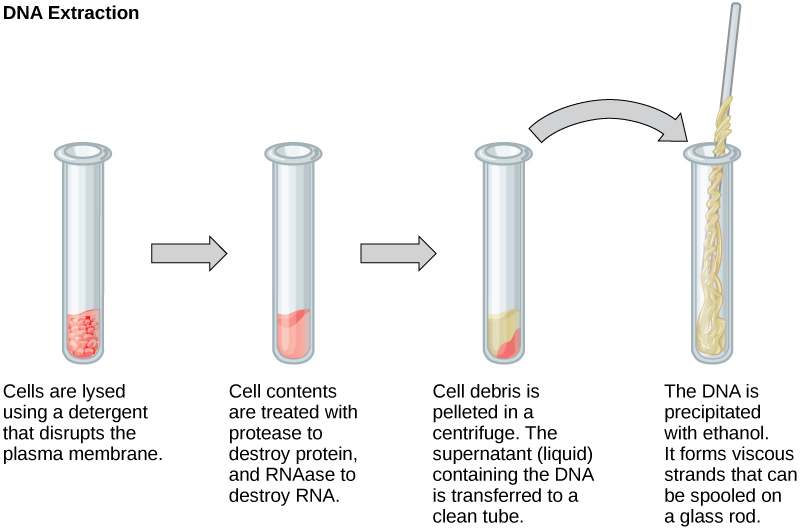
Figure 2. This diagram shows the basic method used for extraction of DNA.
RNA analysis is performed to study gene expression patterns in cells. RNA is naturally very unstable because RNAses are commonly present in nature and very difficult to inactivate. Similar to DNA, RNA extraction involves the use of various buffers and enzymes to inactivate macromolecules and preserve the RNA.

Figure 3. Shown are DNA fragments from seven samples run on a gel, stained with a fluorescent dye, and viewed under UV light. (credit: James Jacob, Tompkins Cortland Community College)
Gel Electrophoresis
Because nucleic acids are negatively charged ions at neutral or basic pH in an aqueous environment, they can be mobilized by an electric field. Gel electrophoresis is a technique used to separate molecules on the basis of size, using this charge. The nucleic acids can be separated as whole chromosomes or fragments. The nucleic acids are loaded into a slot near the negative electrode of a semisolid, porous gel matrix and pushed toward the positive electrode at the opposite end of the gel. Smaller molecules move through the pores in the gel faster than larger molecules; this difference in the rate of migration separates the fragments on the basis of size. There are molecular weight standard samples that can be run alongside the molecules to provide a size comparison. Nucleic acids in a gel matrix can be observed using various fluorescent or colored dyes. Distinct nucleic acid fragments appear as bands at specific distances from the top of the gel (the negative electrode end) on the basis of their size (Figure 3). A mixture of genomic DNA fragments of varying sizes appear as a long smear, whereas uncut genomic DNA is usually too large to run through the gel and forms a single large band at the top of the gel.
Amplification of Nucleic Acid Fragments by Polymerase Chain Reaction
Although genomic DNA is visible to the naked eye when it is extracted in bulk, DNA analysis often requires focusing on one or more specific regions of the genome. Polymerase chain reaction (PCR) is a technique used to amplify specific regions of DNA for further analysis (Figure 4). PCR is used for many purposes in laboratories, such as the cloning of gene fragments to analyze genetic diseases, identification of contaminant foreign DNA in a sample, and the amplification of DNA for sequencing. More practical applications include the determination of paternity and detection of genetic diseases.

Figure 4. Polymerase chain reaction, or PCR, is used to amplify a specific sequence of DNA. Primers—short pieces of DNA complementary to each end of the target sequence—are combined with genomic DNA, Taq polymerase, and deoxynucleotides. Taq polymerase is a DNA polymerase isolated from the thermostable bacterium Thermus aquaticus that is able to withstand the high temperatures used in PCR. Thermus aquaticus grows naturally in the Lower Geyser Basin of Yellowstone National Park. Reverse transcriptase PCR (RT-PCR) is similar to PCR, but cDNA is made from an RNA template before PCR begins.
DNA fragments can also be amplified from an RNA template in a process called reverse transcriptase PCR (RT-PCR). The first step is to recreate the original DNA template strand (called cDNA) by applying DNA nucleotides to the mRNA. This process is called reverse transcription. This requires the presence of an enzyme called reverse transcriptase. After the cDNA is made, regular PCR can be used to amplify it.
Hybridization, Southern Blotting, and Northern Blotting
Nucleic acid samples, such as fragmented genomic DNA and RNA extracts, can be probed for the presence of certain sequences. Short DNA fragments called probes are designed and labeled with radioactive or fluorescent dyes to aid detection. Gel electrophoresis separates the nucleic acid fragments according to their size. The fragments in the gel are then transferred onto a nylon membrane in a procedure called blotting (Figure 5). The nucleic acid fragments that are bound to the surface of the membrane can then be probed with specific radioactively or fluorescently labeled probe sequences. When DNA is transferred to a nylon membrane, the technique is called Southern blotting, and when RNA is transferred to a nylon membrane, it is called northern blotting. Southern blots are used to detect the presence of certain DNA sequences in a given genome, and northern blots are used to detect gene expression. Note that “Southern” blotting is capitalized but no other type of blotting; Southern blotting is named after the scientist who pioneered this technique, Edwin Southern. The other types of blotting were named in reference to the original Southern blot.

Figure 5. Southern blotting is used to find a particular sequence in a sample of DNA. DNA fragments are separated on a gel, transferred to a nylon membrane, and incubated with a DNA probe complementary to the sequence of interest. Northern blotting is similar to Southern blotting, but RNA is run on the gel instead of DNA. In western blotting, proteins are run on a gel and detected using antibodies.
DNA Sequencing
Until the 1990s, the sequencing of DNA (reading the sequence of DNA) was a relatively expensive and long process. Using radiolabeled nucleotides also compounded the problem through safety concerns. With currently available technology and automated machines, the process is cheap, safer, and can be completed in a matter of hours. Fred Sanger developed the sequencing method used for the human genome sequencing project, which is widely used today (Figure 6).

Figure 6. In Frederick Sanger’s dideoxy chain termination method, dye-labeled dideoxynucleotides are used to generate DNA fragments that terminate at different points. The DNA is separated by capillary electrophoresis on the basis of size, and from the order of fragments formed, the DNA sequence can be read. The DNA sequence readout is shown on an electropherogram that is generated by a laser scanner.
The method is known as the dideoxy chain termination method. The sequencing method is based on the use of chain terminators, the dideoxynucleotides (ddNTPs). The dideoxynucleotides, or ddNTPSs, differ from the deoxynucleotides by the lack of a free 3′ OH group on the five-carbon sugar. If a ddNTP is added to a growing a DNA strand, the chain is not extended any further because the free 3′ OH group needed to add another nucleotide is not available. By using a predetermined ratio of deoxyribonucleotides to dideoxynucleotides, it is possible to generate DNA fragments of different sizes.
The DNA sample to be sequenced is denatured or separated into two strands by heating it to high temperatures. The DNA is divided into four tubes in which a primer, DNA polymerase, and all four nucleotides (A, T, G, and C) are added. In addition to each of the four tubes, limited quantities of one of the four dideoxynucleotides are added to each tube respectively. The tubes are labeled as A, T, G, and C according to the ddNTP added. For detection purposes, each of the four dideoxynucleotides carries a different fluorescent label. Chain elongation continues until a fluorescent dideoxy nucleotide is incorporated, after which no further elongation takes place. After the reaction is over, electrophoresis is performed. Even a difference in length of a single base can be detected. The sequence is read from a laser scanner. For his work on DNA sequencing, Sanger received a Nobel Prize in chemistry in 1980.
As already mentioned, gel electrophoresis (Figure 3) is a technique used to separate DNA fragments of different sizes. Usually the gel is made of a chemical called agarose. Agarose powder is added to a buffer and heated. After cooling, the gel solution is poured into a casting tray. Once the gel has solidified, the DNA is loaded on the gel and electric current is applied. The DNA has a net negative charge and moves from the negative electrode toward the positive electrode. The electric current is applied for sufficient time to let the DNA separate according to size; the smallest fragments will be farthest from the well (where the DNA was loaded), and the heavier molecular weight fragments will be closest to the well. Once the DNA is separated, the gel is stained with a DNA-specific dye for viewing it.
Neanderthal Genome: How Are We Related?
The first draft sequence of the Neanderthal genome was published by Richard E. Green et al. in 2010.[1] Neanderthals are the closest ancestors of present-day humans. They were known to have lived in Europe and Western Asia before they disappeared from fossil records approximately 30,000 years ago. Green’s team studied almost 40,000-year-old fossil remains that were selected from sites across the world. Extremely sophisticated means of sample preparation and DNA sequencing were employed because of the fragile nature of the bones and heavy microbial contamination. In their study, the scientists were able to sequence some four billion base pairs. The Neanderthal sequence was compared with that of present-day humans from across the world. After comparing the sequences, the researchers found that the Neanderthal genome had 2 to 3 percent greater similarity to people living outside Africa than to people in Africa. While current theories have suggested that all present-day humans can be traced to a small ancestral population in Africa, the data from the Neanderthal genome may contradict this view. Green and his colleagues also discovered DNA segments among people in Europe and Asia that are more similar to Neanderthal sequences than to other contemporary human sequences. Another interesting observation was that Neanderthals are as closely related to people from Papua New Guinea as to those from China or France. This is surprising because Neanderthal fossil remains have been located only in Europe and West Asia. Most likely, genetic exchange took place between Neanderthals and modern humans as modern humans emerged out of Africa, before the divergence of Europeans, East Asians, and Papua New Guineans.
Several genes seem to have undergone changes from Neanderthals during the evolution of present-day humans. These genes are involved in cranial structure, metabolism, skin morphology, and cognitive development. One of the genes that is of particular interest is RUNX2, which is different in modern day humans and Neanderthals. This gene is responsible for the prominent frontal bone, bell-shaped rib cage, and dental differences seen in Neanderthals. It is speculated that an evolutionary change in RUNX2 was important in the origin of modern-day humans, and this affected the cranium and the upper body.
Watch Svante Pääbo’s talk explaining the Neanderthal genome research at the 2011 annual TED (Technology, Entertainment, Design) conference.
Molecular Cloning
In general, the word “cloning” means the creation of a perfect replica; however, in biology, the re-creation of a whole organism is referred to as “reproductive cloning.” Long before attempts were made to clone an entire organism, researchers learned how to reproduce desired regions or fragments of the genome, a process that is referred to as molecular cloning.
Cloning small fragments of the genome allows for the manipulation and study of specific genes (and their protein products), or noncoding regions in isolation. A plasmid (also called a vector) is a small circular DNA molecule that replicates independently of the chromosomal DNA of microorganisms such as E. coli. In cloning, the plasmid molecules can be used to provide a “folder” in which to insert a desired DNA fragment. Plasmids are usually introduced into a bacterial host for proliferation. In the bacterial context, the fragment of DNA from the human genome (or the genome of another organism that is being studied) is referred to as foreign DNA, or a transgene, to differentiate it from the DNA of the bacterium, which is called the host DNA.
Plasmids occur naturally in bacterial populations (such as Escherichia coli) and have genes that can contribute favorable traits to the organism, such as antibiotic resistance (the ability to be unaffected by antibiotics). Plasmids have been repurposed and engineered as vectors for molecular cloning and the large-scale production of important reagents, such as insulin and human growth hormone. An important feature of plasmid vectors is the ease with which a foreign DNA fragment can be introduced via the multiple cloning site (MCS). The MCS is a short DNA sequence containing multiple sites that can be cut with different commonly available restriction endonucleases. Restriction endonucleases recognize specific DNA sequences and cut them in a predictable manner; they are naturally produced by bacteria as a defense mechanism against foreign DNA. Many restriction endonucleases make staggered cuts in the two strands of DNA, such that the cut ends have a 2- or 4-base single-stranded overhang. Because these overhangs are capable of annealing with complementary overhangs, these are called “sticky ends.” Addition of an enzyme called DNA ligase permanently joins the DNA fragments via phosphodiester bonds. In this way, any DNA fragment generated by restriction endonuclease cleavage can be spliced between the two ends of a plasmid DNA that has been cut with the same restriction endonuclease (Figure 1).
Recombinant DNA Molecules
Plasmids with foreign DNA inserted into them are called recombinant DNA molecules because they are created artificially and do not occur in nature. They are also called chimeric molecules because the origin of different parts of the molecules can be traced back to different species of biological organisms or even to chemical synthesis. Proteins that are expressed from recombinant DNA molecules are called recombinant proteins. Not all recombinant plasmids are capable of expressing genes. The recombinant DNA may need to be moved into a different vector (or host) that is better designed for gene expression. Plasmids may also be engineered to express proteins only when stimulated by certain environmental factors, so that scientists can control the expression of the recombinant proteins.
Cellular Cloning
Unicellular organisms, such as bacteria and yeast, naturally produce clones of themselves when they replicate asexually by binary fission; this is known as cellular cloning. The nuclear DNA duplicates by the process of mitosis, which creates an exact replica of the genetic material.
Practice Question

Figure 1. This diagram shows the steps involved in molecular cloning.
You are working in a molecular biology lab and, unbeknownst to you, your lab partner left the foreign genomic DNA that you are planning to clone on the lab bench overnight instead of storing it in the freezer. As a result, it was degraded by nucleases, but still used in the experiment. The plasmid, on the other hand, is fine. What results would you expect from your molecular cloning experiment?
- There will be no colonies on the bacterial plate.
- There will be blue colonies only.
- There will be blue and white colonies.
- The will be white colonies only.
Reproductive Cloning
Reproductive cloning is a method used to make a clone or an identical copy of an entire multicellular organism. Most multicellular organisms undergo reproduction by sexual means, which involves genetic hybridization of two individuals (parents), making it impossible for generation of an identical copy or a clone of either parent. Recent advances in biotechnology have made it possible to artificially induce asexual reproduction of mammals in the laboratory.
Parthenogenesis, or “virgin birth,” occurs when an embryo grows and develops without the fertilization of the egg occurring; this is a form of asexual reproduction. An example of parthenogenesis occurs in species in which the female lays an egg and if the egg is fertilized, it is a diploid egg and the individual develops into a female; if the egg is not fertilized, it remains a haploid egg and develops into a male. The unfertilized egg is called a parthenogenic, or virgin, egg. Some insects and reptiles lay parthenogenic eggs that can develop into adults.
Sexual reproduction requires two cells; when the haploid egg and sperm cells fuse, a diploid zygote results. The zygote nucleus contains the genetic information to produce a new individual. However, early embryonic development requires the cytoplasmic material contained in the egg cell. This idea forms the basis for reproductive cloning. Therefore, if the haploid nucleus of an egg cell is replaced with a diploid nucleus from the cell of any individual of the same species (called a donor), it will become a zygote that is genetically identical to the donor. Somatic cell nuclear transfer is the technique of transferring a diploid nucleus into an enucleated egg. It can be used for either therapeutic cloning or reproductive cloning.
The first cloned animal was Dolly, a sheep who was born in 1996. The success rate of reproductive cloning at the time was very low. Dolly lived for seven years and died of respiratory complications (Figure 2). There is speculation that because the cell DNA belongs to an older individual, the age of the DNA may affect the life expectancy of a cloned individual. Since Dolly, several animals such as horses, bulls, and goats have been successfully cloned, although these individuals often exhibit facial, limb, and cardiac abnormalities. There have been attempts at producing cloned human embryos as sources of embryonic stem cells, sometimes referred to as cloning for therapeutic purposes. Therapeutic cloning produces stem cells to attempt to remedy detrimental diseases or defects (unlike reproductive cloning, which aims to reproduce an organism). Still, therapeutic cloning efforts have met with resistance because of bioethical considerations.
Practice Question

Figure 2. Dolly the sheep was the first mammal to be cloned. To create Dolly, the nucleus was removed from a donor egg cell. The nucleus from a second sheep was then introduced into the cell, which was allowed to divide to the blastocyst stage before being implanted in a surrogate mother. (credit: modification of work by “Squidonius”/Wikimedia Commons)
Do you think Dolly was a Finn-Dorset or a Scottish Blackface sheep?
Genetic Engineering
Genetic engineering is the alteration of an organism’s genotype using recombinant DNA technology to modify an organism’s DNA to achieve desirable traits. The addition of foreign DNA in the form of recombinant DNA vectors generated by molecular cloning is the most common method of genetic engineering. The organism that receives the recombinant DNA is called a genetically modified organism (GMO). If the foreign DNA that is introduced comes from a different species, the host organism is called transgenic. Bacteria, plants, and animals have been genetically modified since the early 1970s for academic, medical, agricultural, and industrial purposes. In the US, GMOs such as Roundup-ready soybeans and borer-resistant corn are part of many common processed foods.
Gene Targeting
Although classical methods of studying the function of genes began with a given phenotype and determined the genetic basis of that phenotype, modern techniques allow researchers to start at the DNA sequence level and ask: “What does this gene or DNA element do?” This technique, called reverse genetics, has resulted in reversing the classic genetic methodology. This method would be similar to damaging a body part to determine its function. An insect that loses a wing cannot fly, which means that the function of the wing is flight. The classical genetic method would compare insects that cannot fly with insects that can fly, and observe that the non-flying insects have lost wings. Similarly, mutating or deleting genes provides researchers with clues about gene function. The methods used to disable gene function are collectively called gene targeting. Gene targeting is the use of recombinant DNA vectors to alter the expression of a particular gene, either by introducing mutations in a gene, or by eliminating the expression of a certain gene by deleting a part or all of the gene sequence from the genome of an organism.
Check Your Understanding
Answer the question(s) below to see how well you understand the topics covered in the previous section. This short quiz does not count toward your grade in the class, and you can retake it an unlimited number of times.
Use this quiz to check your understanding and decide whether to (1) study the previous section further or (2) move on to the next section.
Candela Citations
- Introduction to Key Technologies. Authored by: Shelli Carter and Lumen Learning. Provided by: Lumen Learning. License: CC BY: Attribution
- Biology. Provided by: OpenStax CNX. Located at: http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@10.8. License: CC BY: Attribution. License Terms: Download for free at http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@10.8
- Svante Paabo: DNA clues to our inner neanderthal. Authored by: TED. Located at: https://youtu.be/kU0ei9ApmsY. License: All Rights Reserved. License Terms: Standard YouTube License
- Richard E. Green et al., “A Draft Sequence of the Neandertal Genome,” Science 328 (2010): 710–22. ↵