Learning Outcomes
- Identify common errors that can create an abnormal karyotype
- Identify syndromes that result from a significant change in chromosome number
Of all of the chromosomal conditions, abnormalities in chromosome number are the most obviously identifiable from a karyogram. Conditions of chromosome number include the duplication or loss of entire chromosomes, as well as changes in the number of complete sets of chromosomes. They are caused by nondisjunction, which occurs when pairs of homologous chromosomes or sister chromatids fail to separate during meiosis. Misaligned or incomplete synapsis, or a dysfunction of the spindle apparatus that facilitates chromosome migration, can cause nondisjunction. The risk of nondisjunction occurring increases with the age of the parents.
Nondisjunction can occur during either meiosis I or II, with differing results (Figure 1). If homologous chromosomes fail to separate during meiosis I, the result is two gametes that lack that particular chromosome and two gametes with two copies of the chromosome. If sister chromatids fail to separate during meiosis II, the result is one gamete that lacks that chromosome, two normal gametes with one copy of the chromosome, and one gamete with two copies of the chromosome.
Practice Question
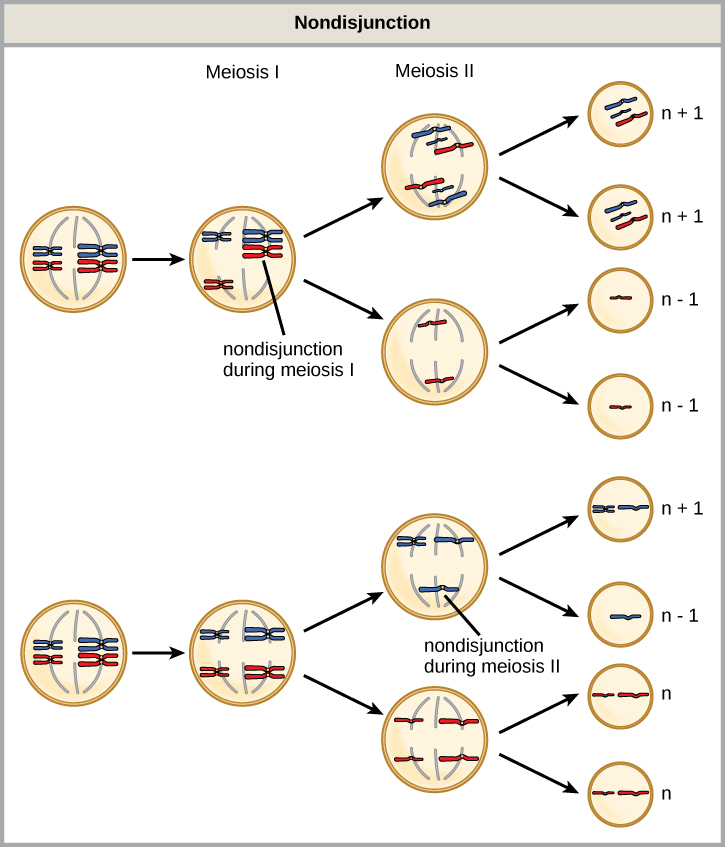
Figure 1. Nondisjunction occurs when homologous chromosomes or sister chromatids fail to separate during meiosis, resulting in an abnormal chromosome number. Nondisjunction may occur during meiosis I or meiosis II.
Which of the following statements about nondisjunction is true?
- Nondisjunction only results in gametes with n+1 or n–1 chromosomes.
- Nondisjunction occurring during meiosis II results in 50 percent normal gametes.
- Nondisjunction during meiosis I results in 50 percent normal gametes.
- Nondisjunction always results in four different kinds of gametes.
Aneuploidy

Figure 2. The incidence of having a fetus with trisomy 21 increases dramatically with maternal age.
An individual with the typical number of chromosomes for their species is called euploid; in humans, euploidy corresponds to 22 pairs of autosomes and one pair of sex chromosomes. An individual with an error in chromosome number is described as aneuploid, a term that includes monosomy (loss of one chromosome) or trisomy (gain of an extraneous chromosome). Monosomic human zygotes missing any one copy of an autosome invariably fail to develop to birth because they lack essential genes. This underscores the importance of “gene dosage” in humans. Most autosomal trisomies also fail to develop to birth; however, duplications of some of the smaller chromosomes (13, 15, 18, 21, or 22) can result in offspring that survive for several weeks to many years. Trisomic individuals experience a different type of genetic imbalance: an excess in gene dose. Individuals with an extra chromosome may synthesize an abundance of the gene products encoded by that chromosome. This extra dose (150 percent) of specific genes can lead to a number of functional challenges and often precludes development. The most common trisomy among viable births is that of chromosome 21, which corresponds to Down Syndrome. Individuals with this inherited condition are characterized by short stature and stunted digits, facial distinctions that include a broad skull and large tongue, and significant developmental delays. The incidence of Down syndrome is correlated with maternal age; older women are more likely to become pregnant with fetuses carrying the trisomy 21 genotype (Figure 2).
Polyploidy

Figure 3. As with many polyploid plants, this triploid orange daylily (Hemerocallis fulva) is particularly large and robust, and grows flowers with triple the number of petals of its diploid counterparts. (credit: Steve Karg)
An individual with more than the typical number of chromosome sets (two for diploid species) is called polyploid. For instance, fertilization of an abnormal diploid egg with a normal haploid sperm would yield a triploid zygote. Polyploid animals are extremely rare, with only a few examples among the flatworms, crustaceans, amphibians, fish, and lizards. Polyploid animals are sterile because meiosis cannot proceed normally and instead produces mostly aneuploid daughter cells that cannot yield viable zygotes. Rarely, polyploid animals can reproduce asexually by haplodiploidy, in which an unfertilized egg divides mitotically to produce offspring. In contrast, polyploidy is very common in the plant kingdom, and polyploid plants tend to be larger and more robust than euploids of their species (Figure 3).
Sex Chromosome Nondisjunction in Humans
Humans display dramatic deleterious effects with autosomal trisomies and monosomies. Therefore, it may seem counterintuitive that human females and males can function normally, despite carrying different numbers of the X chromosome. Rather than a gain or loss of autosomes, variations in the number of sex chromosomes are associated with relatively mild effects. In part, this occurs because of a molecular process called X inactivation. Early in development, when female mammalian embryos consist of just a few thousand cells (relative to trillions in the newborn), one X chromosome in each cell inactivates by tightly condensing into a quiescent (dormant) structure called a Barr body. The chance that an X chromosome (maternally or paternally derived) is inactivated in each cell is random, but once the inactivation occurs, all cells derived from that one will have the same inactive X chromosome or Barr body. By this process, females compensate for their double genetic dose of X chromosome.

Figure 4. In cats, the gene for coat color is located on the X chromosome. In the embryonic development of female cats, one of the two X chromosomes is randomly inactivated in each cell, resulting in a tortoiseshell pattern if the cat has two different alleles for coat color. Male cats, having only one X chromosome, never exhibit a tortoiseshell coat color. (credit: Michael Bodega)
In so-called “tortoiseshell” cats, embryonic X inactivation is observed as color variegation (Figure 4). Females that are heterozygous for an X-linked coat color gene will express one of two different coat colors over different regions of their body, corresponding to whichever X chromosome is inactivated in the embryonic cell progenitor of that region.
An individual carrying an abnormal number of X chromosomes will inactivate all but one X chromosome in each of their cells. However, even inactivated X chromosomes continue to express a few genes, and X chromosomes must reactivate for the proper maturation of ovaries. As a result, X-chromosomal abnormalities are typically associated with mild mental and physical defects, as well as sterility. If the X chromosome is absent altogether, the individual will not develop in utero.
Several errors in sex chromosome number have been characterized. Individuals with three X chromosomes, called triplo-X, are phenotypically female but express developmental delays and reduced fertility. The XXY genotype, corresponding to one type of Klinefelter syndrome, corresponds to phenotypically male individuals with small testes, enlarged breasts, and reduced body hair. More complex types of Klinefelter syndrome exist in which the individual has as many as five X chromosomes. In all types, every X chromosome except one undergoes inactivation to compensate for the excess genetic dosage. This can be seen as several Barr bodies in each cell nucleus. Turner syndrome, characterized as an X0 genotype (i.e., only a single sex chromosome), corresponds to a phenotypically female individual with short stature, webbed skin in the neck region, hearing and cardiac impairments, and sterility.
Duplications and Deletions
In addition to the loss or gain of an entire chromosome, a chromosomal segment may be duplicated or lost. Duplications and deletions often produce offspring that survive but exhibit physical and mental abnormalities. Duplicated chromosomal segments may fuse to existing chromosomes or may be free in the nucleus. Cri-du-chat (from the French for “cry of the cat”) is a syndrome associated with nervous system abnormalities and identifiable physical features that result from a deletion of most of 5p (the small arm of chromosome 5) (Figure 5). Infants with this genotype emit a characteristic high-pitched cry on which the condition’s name is based.

Figure 5. This individual with cri-du-chat syndrome is shown at two, four, nine, and 12 years of age. (credit: Paola Cerruti Mainardi)
Try It
Candela Citations
- Biology 2e. Provided by: OpenStax. Located at: http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@10.8. License: CC BY: Attribution. License Terms: Access for free at https://openstax.org/books/biology-2e/pages/1-introduction
