Introduction
The Brønsted-Lowry acid-base theory is still used extensively, but it is very restrictive as it focuses primarily on acids and bases acting as proton donors and acceptors. Sometimes conditions arise where the theory doesn’t necessarily fit, such as in solids and gases. In 1923, G.N. Lewis from UC Berkeley proposed an alternate theory to describe acids and bases. His theory gave a generalized explanation of acids and bases based on structure and bonding. Through the use of the Lewis definition of acids and bases, chemists are now able to predict a wider variety of acid-base reactions. Lewis’ theory used electrons instead of proton transfer and specifically stated that an acid is a species that accepts an electron pair while a base donates an electron pair.

Figure 1: Above: A Lewis Base (B) donates it electrons to a Lewis Acid (A) resulting in a coordinate covalently bonded compound, also known as an adduct.
The reaction of a Lewis acid and a Lewis base will produce a coordinate covalent bond, as shown in Figure 1 above. A coordinate covalent bond is just a type of covalent bond in which one reactant gives it electron pair to another reactant. In this case the Lewis base donates its electrons to the Lewis acid. When they do react this way the resulting product is called an adduct.
- Lewis acid: a species that accepts an electron pair (i.e., an electrophile) and will have vacant orbitals
- Lewis base: a species that donates an electron pair (i.e., a nucleophile) and will have lone-pair electrons
Lewis acids
Lewis acids accept an electron pair. Lewis acids are electrophilic meaning that they are electron attracting. When bonding with a base the acid uses its lowest unoccupied molecular orbital or LUMO (see figure).
- Various species can act as Lewis acids. All cations are Lewis acids since they are able to accept electrons. (e.g., Cu2+, Fe2+, Fe3+)
- An atom, ion, or molecule with an incomplete octet of electrons can act as an Lewis acid (e.g., BF3, AlF3).
- Molecules where the central atom can have more than 8 valence shell electrons can be electron acceptors, and thus are classified as Lewis acids (e.g., SiBr4, PF5).
- Molecules that have multiple bonds between two atoms of different electronegativities (e.g., CO2, SO2)
Lewis bases
Lewis bases donate an electron pair. Lewis bases are nucleophilic meaning that they “attack” a positive charge with their lone pair. They utilize the highest occupied molecular orbital or HOMO (Figure 2). An atom, ion, or molecule with a lone-pair of electrons can thus be a Lewis base. Each of the following anions can “give up” their electrons to an acid, e.g., $$OH^-$$, $$CN^-$$, $$CH_3COO^-$$, $$:NH_3$$, $$H_2O:$$, $$CO:$$. The Lewis base’s HOMO (highest occupied molecular orbital) interacts with the Lewis acid’s LUMO (lowest unoccupied molecular orbital) to create bonded molecular orbitals. Both Lewis acids and bases contain HOMO and LUMOs but only the HOMO is considered for bases and only the LUMO is considered for acids (see figure).

Lewis acids have vacant orbitals so they are in a lower energy level, while Lewis bases have lone pair electrons to share and thus occupy a higher energy level.
Amphoterism
As of now you should know that acids and bases are distinguished as two separate things however some substances can be both an acid and a base. You may have noticed this with water, which can act as both an acid or a base. This ability of water to do this makes it an amphoteric molecule. Water can act as an acid by donating its proton to the base and thus becoming its conjugate acid, OH-. However, water can also act as a base by accepting a proton from an acid to become its conjugate base, H3O+.
- Water acting as an acid:
\[H_2O + NH_3 \rightarrow NH_4^+ + OH^- \label{3}\]
- Water acting as a base:
\[H_2O + HCl \rightarrow Cl^- + H_3O^+ \label{4}\]
You may have noticed that the degree to which a molecule acts depends on the medium in which the molecule has been placed in. Water does not act as an acid in an acid medium and does not act as a base in a basic medium. Thus, the medium which a molecule is placed in has an effect on the properties of that molecule. Other molecules can also act as either an acid or a base.
The interaction between a magnesium cation (Mg2+) and a carbonyl oxygen is a common example of a Lewis acid-base reaction in enzyme-catalyzed biological reactions. The carbonyl oxygen (the Lewis base) donates a pair of electrons to the magnesium cation (the Lewis acid).

Further reading
References
- Cycloaddition on Ge(100) of the Lewis Acid AlCl3. Soon Jung Jung,, Young-Sang Youn,, Hangil Lee,,, Ki-Jeong Kim,,, Bong Soo Kim, and, Sehun Kim,. Journal of the American Chemical Society 2008 130 (11), 3288-3289
- Fluorescence Maxima of 10-Methylacridone-Metal Ion Salt Complexes: A Convenient and Quantitative Measure of Lewis Acidity of Metal Ion Salts. Shunichi Fukuzumi and Kei Ohkubo. Journal of the American Chemical Society 2002 124 (35), 10270-10271.
- Harwood, William S., F. G. Herring, Jeffry D. Madura, and Ralph H. Petrucci. General Chemistry Principles and Modern Applications. 9th ed. New Jersey: Prentice Hall, 2007. 695-96.
Nucleophiles and electrophiles
The majority of organic reactions can in fact be classified as Lewis acid-base reactions. However, organic chemists usually refer to a Lewis acid as an electrophile (which is electron poor), and a Lewis base as a nucleophile (electron rich). The pair of electrons in a mechanistic curved arrow begins at a nucleophilic center and end at an electrophilic center. To draw a mechanism, we need to think about which atom is the nucleophile – usually giving away a lone pair or a bond – and which atom is the electrophile – gaining a lone pair or bond.
Just as with Bronsted-Lowry acids/bases, we can have strong Lewis acids (electrophiles) and strong Lewis bases (nucleophiles). However, whereas with Bronsted-Lowry we can quantify acid/base strength using the pKa/pKb, this is not possible with electrophiles & nucleophiles. This is because electrophile/nucleophile strength is affected by multiple factors, and involve nearly all the chemical elements (not just hydrogen).
For organic chemistry, though, we will mainly be focused on reactions at carbon, so it is useful to discuss nucleophile/electrophile strength in these terms. To some extent, we will find parallels between the two acid-base theories, so that a strong Bronsted-Lowry base will often be a good nucleophile, and a strong Bronsted-Lowry acid will often be a good electrophile. In other words, the same quality of ‘electron-richness’ that makes a something nucleophilic also makes it Bronsted-Lowry basic. But carbon is a more polarizable atom (often said to be “softer”) than hydrogen, and this leads to some marked differences. For example, iodide ion (I–) is a very weak Bronsted-Lowry base but a strong nucleophile towards carbon; hydride ion (H–) is a strong Bronsted-Lowry base but a poor nucleophile towards carbon. When we look at these Lewis acid-base reactions, we will need to judge nucleophile/electrophile strength slightly differently than we did for Bronsted-Lowry reactions, though many factors remain similar.
What makes a nucleophile?
A common way to examine nucleophile strength involves looking at nucleophilic substitution reactions, in particular the SN2 reaction that we will study in detail in chapter 8. This is where a nucleophile attacks a carbon, forming a new bond, while another group (the “leaving group”) departs from the carbon on the other side. The electron flow can be seen as similar to a Bronsted-Lowry acid-base reaction, but with a carbon center being transferred (from Cl to O) instead of a hydrogen. Remember, the nucleophile acts as the (Lewis) base.
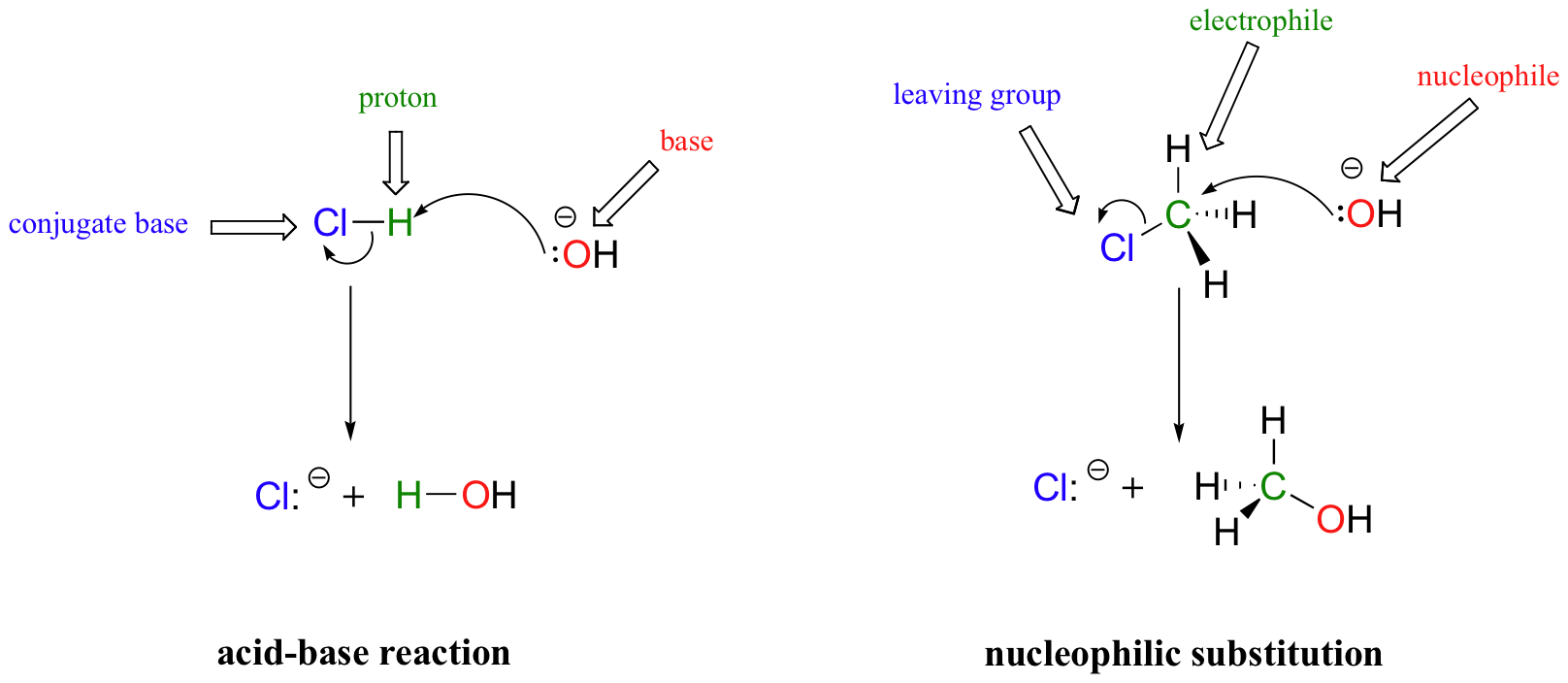
In both reaction types, we are looking at very similar players: an electron-rich species (the nucleophile/base) attacks an electron-poor species (the electrophile/proton), driving off the leaving group/conjugate base.
As a general rule, nucleophile substitution reactions that involve powerful nucleophiles tend to occur with SN2 mechanisms, while reactions with weaker nucleophiles tend to go via a multistep mechanism (called SN1). But what makes a group a strong or weak nucleophile? Nucleophilic functional groups are those which have electron-rich atoms able to donate a pair of electrons to form a new covalent bond. In both laboratory and biological organic chemistry, the most relevant nucleophilic atoms are oxygen, nitrogen, and sulfur, and the most common nucleophilic functional groups are water, alcohols, phenols, amines, thiols, and occasionally carboxylates.
More specifically in laboratory reactions, halide and azide (N3–) anions are commonly seen acting as nucleophiles.
Of course, carbons can also be nucleophiles – otherwise how could new carbon-carbon bonds be formed in the synthesis of large organic molecules like DNA or fatty acids? Enolate ions are the most common carbon nucleophiles in biochemical reactions, while the cyanide ion (CN–) is just one example of a carbon nucleophile commonly used in the laboratory. Reactions with nucleophiles will be covered in detail in chapters 8 and 9.
Nucleophile strength
Now, let’s discuss some of the major factors that affect nucleophile strength or “nucleophilicity”. First, you should realize that a strong nucleophile is a reactive or unstable nucleophile; one that is stable will be weak and unreactive. That means factors that stabilize a nucleophile will make it weaker.
Charge and nucleophilicity
The charge on a nucleophilic atom has a very large effect on its nucleophilicity. This is an idea that makes intuitive sense: a hydroxide ion is much more nucleophilic (and basic) than a water molecule, because the negatively charged oxygen on the hydroxide ion carries greater electron density than the oxygen atom of a neutral water molecule. In practical terms, this means that a hydroxide nucleophile will react in an SN2 reaction with bromomethane much faster ( about 10,000 times faster) than a water nucleophile.
A neutral amine is nucleophilic, whereas a protonated ammonium cation is not. This is why enzymes which have evolved to catalyze nucleophilic reactions often have a basic amino acid side chain poised in position to accept a proton from the nucleophilic atom as the nucleophilic attack occurs.

Depending on the specific reaction being discussed, deprotonation of the nucleophile might occur before, during, or after the actual nucleophilic attack.
Periodic trends and solvent effects in nucleophilicity
There are predictable periodic trends in nucleophilicity. Moving horizontally across the second row of the table, the trend in nucleophilicity parallels the trend in basicity:

The reasoning behind the horizontal nucleophilicity trend is the same as the reasoning behind the basicity trend: more electronegative elements hold their electrons more tightly, and are less able to donate them to form a new bond.
This horizontal trends also tells us that amines are more nucleophilic than alcohols, although both groups commonly act as nucleophiles in both laboratory and biochemical reactions.
Recall from the previous section that the basicity of atoms decreases as we move vertically down a column on the periodic table: –SR (thiolate) ions are less basic than –OR (alkoxide ions), for example, and bromide ion (Br–) is less basic than chloride ion (Cl–), which in turn is less basic than fluoride ion (F–). Recall also that this trend can be explained by considering the increasing size of the ‘electron cloud’ around the larger ions: the electron density inherent in the negative charge is spread around a larger area, which tends to increase stability (and thus reduce basicity).
The vertical periodic trend for nucleophilicity is somewhat more complicated that that for basicity: depending on the solvent that the reaction is taking place in, the nucleophilicity trend can go in either direction. Let’s take the simple example of the SN2 reaction below:

. . .where Nu– is one of the halide ions: fluoride, chloride, bromide, or iodide, and the leaving group I* is a radioactive isotope of iodine (which allows us to distinguish the leaving group from the nucleophile in that case where both are iodide). If this reaction is occurring in a protic solvent (that is, a solvent that has a hydrogen bonded to an oxygen or nitrogen – water, methanol and ethanol are the most important examples), then the reaction will go fastest when iodide is the nucleophile, and slowest when fluoride is the nucleophile, reflecting the relative strength of the nucleophile.

Relative nucleophilicity in a protic solvent
This of course, is opposite that of the vertical periodic trend for basicity, where iodide is the least basic (you may want to review the reasoning for this trend in section 7.3A). What is going on here? Shouldn’t the stronger base, with its more reactive unbonded valence electrons, also be the stronger nucleophile?
As mentioned above, it all has to do with the solvent. Remember, we are talking now about the reaction running in a protic solvent like ethanol. Protic solvent molecules form very strong ion-dipole interactions with the negatively-charged nucleophile, essentially creating a ‘solvent cage’ around the nucleophile:

In order for the nucleophile to attack the electrophile, it must break free, at least in part, from its solvent cage. The lone pair electrons on the larger, less basic iodide ion interact less tightly with the protons on the protic solvent molecules – thus the iodide nucleophile is better able to break free from its solvent cage compared the smaller, more basic fluoride ion, whose lone pair electrons are bound more tightly to the protons of the cage.
The picture changes if we switch to a polar aprotic solvent, such as acetone, in which there is a molecular dipole but no hydrogens bound to oxygen or nitrogen. Now, fluoride is the best nucleophile, and iodide the weakest.

Relative nucleophilicity in a polar aprotic solvent
The reason for the reversal is that, with an aprotic solvent, the ion-dipole interactions between solvent and nucleophile are much weaker: the positive end of the solvent’s dipole is hidden in the interior of the molecule, and thus it is shielded from the negative charge of the nucleophile.

A weaker solvent-nucleophile interaction means a weaker solvent cage for the nucleophile to break through, so the solvent effect is much less important, and the more basic fluoride ion is also the better nucleophile.
Why not use a completely nonpolar solvent, such as hexane, for this reaction, so that the solvent cage is eliminated completely? The answer to this is simple – the nucleophile needs to be in solution in order to react at an appreciable rate with the electrophile, and a solvent such as hexane will not solvate an a charged (or highly polar) nucleophile at all. That is why chemists use polar aprotic solvents for nucleophilic substitution reactions in the laboratory: they are polar enough to solvate the nucleophile, but not so polar as to lock it away in an impenetrable solvent cage. In addition to acetone, three other commonly used polar aprotic solvents are acetonitrile, dimethylformamide (DMF), and dimethyl sulfoxide (DMSO).

In biological chemistry, where the solvent is protic (water), the most important implication of the periodic trends in nucleophilicity is that thiols (RSH) are more powerful nucleophiles than alcohols (ROH). The thiol group in a cysteine amino acid, for example, is a powerful nucleophile and often acts as a nucleophile in enzymatic reactions, and of course negatively-charged thiolates (RS–) are even more nucleophilic. This is not to say that the hydroxyl groups on serine, threonine, and tyrosine do not also act as nucleophiles – they do.
Resonance effects on nucleophilicity
Resonance also affects the strength of the nucleophile. The reasoning involved is the same as that which we used to understand resonance effects on basicity (see section 6.4). If the electron lone pair on a heteroatom is delocalized by resonance, it is inherently less reactive – meaning less nucleophilic, and also less basic. An alkoxide ion, for example, is more nucleophilic and more basic than a carboxylate group (see figure), even though in both cases the nucleophilic atom is a negatively charged oxygen. In the alkoxide, the negative charge is localized on a single oxygen, while in the carboxylate the charge is delocalized over two oxygen atoms by resonance.

The nitrogen atom on an amide is less nucleophilic than the nitrogen of an amine, due to the resonance stabilization of the nitrogen lone pair provided by the amide carbonyl group.

Steric effects on nucleophile strength
Steric hindrance is an important consideration when evaluating nucleophilicity. For example, tert-butanol is less potent as a nucleophile than methanol. This is because the comparatively bulky methyl groups on the tertiary alcohol effectively block the route of attack by the nucleophilic oxygen, slowing the reaction down considerably (imagine trying to walk through a narrow doorway while carrying three large suitcases!).

It is not surprising that it is more common to observe serines acting as nucleophiles in enzymatic reactions compared to threonines – the former is a primary alcohol, while the latter is a secondary alcohol and therefore more hindered.
nucleophile strength
The main factors affecting nucleophile strength are as follows:
- Charge – negatively charged => stronger nucleophile
- Within a row – more electronegative atom => weaker nucleophile
- Within a column, size of atom. Polar protic solvent, bigger atom is better; polar aprotic solvent, smaller atom is better.
- Resonance – if the nucleophilic lone pair can be delocalized by resonance, it will make it less nucleophilic
- Steric hindrance – a hindered nucleophilic atom will tend to be less reactive, particularly when attacking a crowded electrophile.
Examples
In each of the following pairs of molecules/ions, which is the better nucleophile in a reaction with CH3Br in acetone solvent? Explain your choice.
phenolate ion (PhO-) or benzoate ion (deprotonated benzoic acid, PhCOO–)
water and hydronium ion (H3O+)
trimethylamine (Me3N) and triethylamine (Et3N)
chloride anion and iodide anion
CH3NH– and CH3CH2NH2
Electrophiles and carbocation stability
In the vast majority of the nucleophilic substitution reactions you will see in this and other organic chemistry texts, the electrophilic atom is a carbon which is bonded to an electronegative atom, usually oxygen, nitrogen, sulfur, or a halogen. The concept of electrophilicity is relatively simple: an electron-poor atom is an attractive target for something that is electron-rich, i.e. a nucleophile. However, we must also consider the effect of steric hindrance on electrophilicity. In addition, we must discuss how the nature of the electrophilic carbon, and more specifically the stability of a potential carbocationic intermediate, influences the SN1 vs. SN2 character of a nucleophilic substitution reaction. We also need to consider other reactions which do not involve nucleophilic substitution, such as electrophilic addition (chapter 10).
Steric effects on electrophilicity
Consider two hypothetical SN2 reactions: one in which the electrophile is a methyl carbon and another in which it is tertiary carbon.
Because the three substituents on the methyl carbon electrophile are tiny hydrogens, the nucleophile has a relatively clear path for backside attack. However, backside attack on the tertiary carbon is blocked by

the bulkier methyl groups. Once again, steric hindrance – this time caused by bulky groups attached to the electrophile rather than to the nucleophile – hinders the progress of an associative nucleophilic (SN2) displacement.
The factors discussed in the above paragraph, however, do not prevent a sterically-hindered carbon from being a good electrophile – they only make it less likely to be attacked in a concerted SN2 reaction. Nucleophilic substitution reactions in which the electrophilic carbon is sterically hindered are more likely to occur by a two-step, dissociative (SN1) mechanism. This makes perfect sense from a geometric point of view: the limitations imposed by sterics are significant mainly in an SN2 displacement, when the electrophile being attacked is a sp3-hybridized tetrahedral carbon with its relatively ‘tight’ angles of 109.4o. In an SN1 mechanism, the nucleophile attacks an sp2-hybridized carbocation intermediate, which has trigonal planar geometry with ‘open’ 120o angles.

With this open geometry, the empty p orbital of the electrophilic carbocation is no longer significantly shielded from the approaching nucleophile by the bulky alkyl groups. A carbocation is a very potent electrophile, and the nucleophilic step occurs very rapidly compared to the first (ionization) step.
Stability of carbocation intermediates
There are several common mechanisms where the rate-limiting step is the formation of a carbocation intermediate, which is an important electrophile for us to consider. The rate of this step – and therefore, the rate of the overall reaction – depends on the activation energy for the process where the carbocation forms. According to Hammond’s postulate (section 5.5.), the more stable the carbocation intermediate is, the faster this step will be. In other words, the rate of a carbocation-based mechanism depends to a large degree on the stability of the carbocation intermediate that forms.
The critical question now becomes, “what stabilizes a carbocation?” Think back to section 6.4., when we were learning how to evaluate the strength of an acid. The critical question was “how stable is the conjugate base that results when this acid donates its proton”? In many cases, this conjugate base was an anion – a center of excess electron density. Anything that can draw some of this electron density away– in other words, any electron withdrawing group – will stabilize the anion.
So if it takes an electron withdrawing group to stabilize a negative charge, what will stabilize a positive charge? An electron donating group!

A positively charged species such as a carbocation is very electron-poor, and thus anything which donates electron density to the center of electron poverty will help to stabilize it. Conversely, a carbocation will be destabilized by an electron withdrawing group.
As we observed in section 5.6., Alkyl groups – methyl, ethyl, and the like – are weak electron donating groups, and thus stabilize nearby carbocations. What this means is that, in general, more substituted carbocations are more stable: a tert-butyl carbocation, for example, is more stable than an isopropyl carbocation. Primary carbocations are highly unstable and not often observed as reaction intermediates; methyl carbocations are even less stable.

Alkyl groups are electron donating and carbocation-stabilizing because the electrons around the neighboring carbons are drawn towards the nearby positive charge, thus slightly reducing the electron poverty of the positively-charged carbon.
It is not accurate to say, however, that carbocations with higher substitution are always more stable than those with less substitution. Just as electron-donating groups can stabilize a carbocation, electron-withdrawing groups act to destabilize carbocations. Carbonyl groups are electron-withdrawing by inductive effects, due to the polarity of the C=O double bond. It is possible to demonstrate in the laboratory that carbocation A below is more stable than carbocation B, even though A is a primary carbocation and B is secondary.

The difference in stability can be explained by considering the electron-withdrawing inductive effect of the ester carbonyl. Recall that inductive effects – whether electron-withdrawing or donating – are relayed through covalent bonds and that the strength of the effect decreases rapidly as the number of intermediary bonds increases. In other words, the effect decreases with distance. In species B the positive charge is closer to the carbonyl group, thus the destabilizing electron-withdrawing effect is stronger than it is in species A.
Stabilization of a carbocation can also occur through resonance effects, and as we have already discussed in the acid-base chapter, resonance effects as a rule are more powerful than inductive effects. Consider the simple case of a benzylic carbocation:

This carbocation is comparatively stable. In this case, electron donation is a resonance effect involving “type IV” resonance. Three additional resonance structures can be drawn for this carbocation in which the positive charge is located on one of three aromatic carbons. The positive charge is not isolated on the benzylic carbon, rather it is delocalized around the aromatic structure: this delocalization of charge results in significant stabilization. As a result, benzylic and allylic carbocations (where the positively charged carbon is conjugated to one or more non-aromatic double bonds) are significantly more stable than even tertiary alkyl carbocations.

Because heteroatoms such as oxygen and nitrogen are more electronegative than carbon, you might expect that they would by definition be electron withdrawing groups that destabilize carbocations. In fact, the opposite is often true: if the oxygen or nitrogen atom is in the correct position, the overall effect is carbocation stabilization. This is due to the fact that although these heteroatoms are electron withdrawing groups by induction, they are electron donating groups via “type III” resonance, and it is this resonance effect which is more powerful. Consider the two pairs of carbocation species below:


In the more stable carbocations, the heteroatom acts as an electron donating group by type III resonance: in effect, the lone pair on the heteroatom is available to delocalize the positive charge. In the less stable carbocations the positively-charged carbon is more than one bond away from the heteroatom, and thus no resonance effects are possible. In fact, in these carbocation species the heteroatoms actually destabilize the positive charge, because they are electron withdrawing by induction.
Finally, vinylic carbocations, in which the positive charge resides on a double-bonded carbon, are very unstable and thus much less likely to form as intermediates in any reaction.
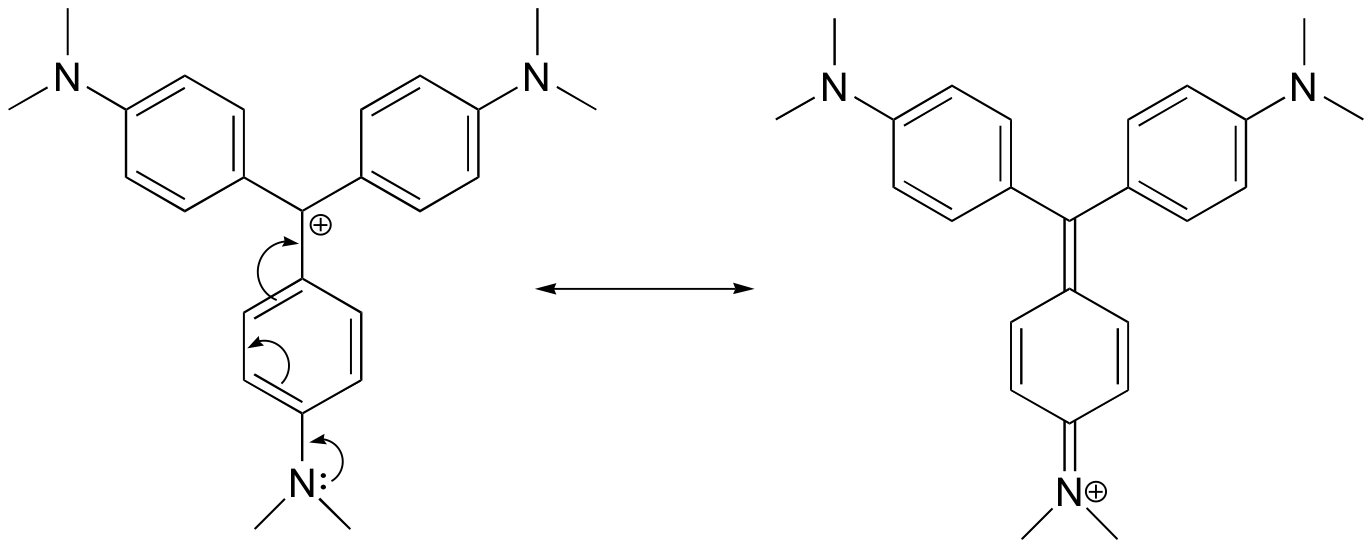
 For the most part, carbocations are very high-energy, transient intermediate species in organic reactions. However, there are some unusual examples of very stable carbocations that take the form of organic salts. Crystal violet is the common name for the chloride salt of the carbocation whose structure is shown below. Notice the structural possibilities for extensive resonance delocalization of the positive charge, and the presence of three electron-donating amine groups.
For the most part, carbocations are very high-energy, transient intermediate species in organic reactions. However, there are some unusual examples of very stable carbocations that take the form of organic salts. Crystal violet is the common name for the chloride salt of the carbocation whose structure is shown below. Notice the structural possibilities for extensive resonance delocalization of the positive charge, and the presence of three electron-donating amine groups.

| Example |
|---|
| Draw a resonance structure of the crystal violet cation in which the positive charge is delocalized to one of the nitrogen atoms.
Show Solution
|
When considering the possibility that a nucleophilic substitution reaction proceeds via an SN1 pathway, it is critical to evaluate the stability of the hypothetical carbocation intermediate. If this intermediate is not sufficiently stable, an SN1 mechanism must be considered unlikely, and the reaction probably proceeds by an SN2 mechanism.
| Example |
|---|
| State which carbocation in each pair below is more stable, or if they are expected to be approximately equal. Explain your reasoning.
Show Solution
|

Candela Citations
- Various. Authored by: Martin A. Walker. Provided by: SUNY Potsdam. Located at: http://directory.potsdam.edu/index.pl?function=user=walkerma. License: CC BY-SA: Attribution-ShareAlike
- Lewis Concept of Acids and Bases. Authored by: Adam Abudra (UCD), Tajinder Badial (UCD). Located at: https://chem.libretexts.org/Textbook_Maps/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Acids_and_Bases/Acid/Lewis_Concept_of_Acids_and_Bases. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 8.3: More about nucleophiles. Authored by: Tim Soderbergu00a0(University of Minnesota, Morris). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/Chapter_08%3A_Nucleophilic_substitution_reactions_I/8.3%3A_More_about_nucleophiles. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 8.4: Electrophiles and carbocation stability. Authored by: Tim Soderberg Associate Professor (Chemistry) at University of Minnesota Morris. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/Chapter_08%3A_Nucleophilic_substitution_reactions_I/8.4%3A_Electrophiles_and_carbocation_stability. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike