This appendix lists most, but not all, reactions covered in this textbook (part 1 only). The focus is on reactions that are most useful in synthesis.
Appendix 1.1. Nucleophilic substitutions with alkyl halides
Overview
Most nucleophilic substitutions used in synthesis go via an SN2 mechanism.
![]()
- SN2 reactions work best with R–X as an alkyl halide or sulfonate. Epoxides also work well for more complex targets.
- Best with 1o alkyl, OK with 2o, fails with 3o and aryl
- Works well with I, Br, OK with Cl, fails with F.
- It requires a strong nucleophile, which usually means one bearing a negative charge such as ¯OH .
- Works well in polar aprotic solvents such as DMSO.
A. Formation of alkyl halides
See section 9.3. Most SN2 reactions utilize alkyl halides, so you need to know how to make them! The most common route is from the corresponding alcohol, using PX3. I recommend using alkyl bromides in synthesis, and these are made using PBr3.
- Use PX3 (X = Cl, Br, I) to convert primary and secondary ROH to RX
- SOCl2 also good for ROH to RCl
- Mechanism involves activation of OH by PX3 to make it a good leaving group, then SN2 attack by X¯.

For example, (2R)-butan-2-ol reacts with PBr3 produces (2S)-2-bromobutane with inversion; note that all three bromines can be delivered.

For preparation of tertiary alkyl chlorides and bromides from the corresponding alcohols, HCl or HBr is the most effective reagent.
B. Formation of alcohols
This is synthetically the reverse of the PBr3 reaction. Here, the nucleophile is hydroxide ion (¯OH). As always with anions, it has to have a cation with it, usually sodium or potassium.
![]()
For example:

C. Formation of amines
See section 9.4.
- NH3, R’NH2 and R’2NH are strong enough to react with R–X, to produce products containing a new R–N bond.
- An excess of the NH3 or amine reactant is needed to suppress the reaction of the product with a second mole of R–X.

There are also indirect methods for preparing amines, which avoid the need for a large excess of amine. One of the most popular uses azide (N3–) as nucleophile, followed by reduction to the amine.

D. Williamson ether synthesis


E. Enolate alkylation
- The enolate nucleophile is usually the conjugate base of a ketone or ester.
- As with the Williamson ether synthesis, the nucleophile has to be made from the ketone or ester with a strong base, usually LDA. (NaH sometimes works, but LDA is usually better.). At -78 oC, LDA forms the less substituted enolate.
- This reaction makes a new C–C bond – often important in synthesis. The reaction works well with primary alkyl bromides or iodides, and moderately with secondary, and fails completely with tertiary alkyl bromides or iodides.

F. Alkyne/Acetylide alkylation
See section 9.8.
- The nucleophile is the conjugate base of an alkyne, called an acetylide.
- Similar to D and E above, the nucleophile has to be made from the alkyne and a strong base, usually NaH or BuLi, though LDA also works.
- As with E above, this makes a new C–C bond.
- Reaction only works well with 1o R–X, since the acetylide is a strong base too.

G. Reduction of R–X with LiAlH4
- LiAlH4 is a strong reducing agent, which in effect delivers hydride ion, H¯. (Hydride ion itself is too small to react well in a direct SN2).
- Reaction only works well with 1o R–X.

Appendix 1.2. Nucleophilic substitutions with Epoxides
H. Reaction of epoxides with strong nucleophiles
- Epoxides are strained 3-membered ring ethers that open very easily with strong nucleophiles, giving alcohols with the former nucleophile attached to the neighboring (less substituted) carbon.
- The mechanism is SN2, with the leaving being the O¯ rather than a halide.
- Any of the above strong nucleophiles (NH3, R’NH2, R’2NH, ¯OH, ¯OR’, enolates, acetylides, LiAlH4, will attack epoxides at the less substituted carbon. For example:

I. Reaction of epoxides with weak nucleophiles
- Epoxides do not open with weak nucleophiles, unless the epoxide is activated first with an acid such as H2SO4.
- The product is an alcohol on the less substituted carbon, with the former nucleophile attached to the neighboring (more substituted) carbon.
- Even at a tertiary center, the mechanism is SN2, with the leaving group being the OH rather than a halide.
- Any weak nucleophiles (H2O, ROH, Cl¯) will attack epoxides at the more substituted carbon, since this is the more electrophilic carbon. For example:

Appendix 1.3. Elimination reactions
Overview
- There are two main mechanisms – E2 and E1.
- E2 reactions make alkenes from alkyl halides and strong base
- E1 reactions are usually used to make alkenes from an alcohol and catalytic acid (H2SO4 or H3PO4).
- E2 is usually better for synthesis, to avoid rearrangements, but alcohol dehydration often works OK.
- Both types of reaction usually obey Zaitsev’s rule – the most substituted/conjugated alkene is the major product.
- All elimination reactions are favored by heat.
J. Formation of alkenes from alkyl halides (E2)
- A strong base is needed, such as KOH, NaOCH3, or KOtBu.
- Zaitsev’s Rule is followed, unless a sterically hindered base such as KOtBu is used.
- Works well with all types of alkyl halide

K. Formation of alkenes from alcohols via dehydration (E1)
- Uses an acid (H2SO4 or H3PO4) to protonate the OH and make it a good leaving group.
- Obeys Zaitsev’s Rule.
- Works best with 3o and 2o but 1o will often work too.

Appendix 1.4. Electrophilic additions to alkenes
Overview
Markovnikov’s Rule
L. Addition of HCl or HBr to alkenes
Addition obeys Markovnikov’s Rule, and forms an alkyl halide with the halogen on the more substituted carbon.

M. Addition of H2O using H3O+
- H2O itself isn’t electrophilic enough to add, but strong acids can add
- The reaction is usually done in two steps, with H2SO4 followed by an H2O quench
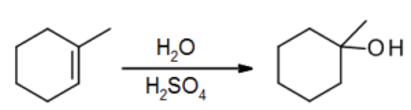
N. Addition of carbenes: Making cyclopropanes
- Carbenes are made from diazo compounds such as diazomethane, by loss of N2 using light.
- Reaction produces a new three-membered ring


O. Addition of Cl2 or Br2
- Initial syn-addition forms a chloronium or bromonium ion with a three-membered ring.
- The halide ion present immediately does SN2 back-side attack to gives the product an anti-dihalide

P. Epoxidation
- Use MCPBA with the alkene
- Product is the syn-epoxide (oxirane) which contains a three-membered ring ether.
- Mechanism involves only a single step.

Q. Oxymercuration-demercuration
- This reaction gives the same alcohol product as with hydration, but the addition is exclusively anti.
- Obeys Markovnikov’s Rule – OH goes on the more substituted carbon.
- Mechanism involves a three-membered ring mercurinium ion intermediate.

R. Anti-Markovnikov addition of water

S. Anti-Markovnikov addition of HBr

Appendix 1.5. Electrophilic additions to alkynes
Alkynes do many of the same reactions as alkenes. However, we will just focus on three main ones.
T. Addition of HCl, HBr or HI
Compare with reaction L above. See section 10.5.
- The reaction follows Markovnikov’s Rule.
-
It can be done with either one or two moles of HX per mole of alkyne, to give different products.
-
When one mole HX is added to an internal alkyne, the product is mainly anti (with Z double bond).
-
If two HX are added, the X atoms both end up on the same (more substituted) carbon.

U. Addition of water to alkynes
Compare with reaction M above. See section 10.5.
- Markovnikov’s Rule applies.
- As with alkenes, an acid catalyst is needed. Internal alkynes can add water under similar conditions to alkenes – H2SO4/H2O.
- Terminal alkynes require a mercury(II) catalyst, usually HgSO4. Unlike oxymercuration, NaBH4 is not needed for forming the product.
- Although an enol is initially formed, this rearranges easily to give a ketone as the isolated product.

V. Anti-Markovnikov addition of H2O to alkynes
- With alkenes, hydroboration-oxidation using BH3.THF produces net anti-Markovnikov hydration; with alkynes, a sterically hindered derivative of BH3 is used, such as dicyclohexylborane (Cy2BH) or disiamylborane (Sia2BH).
- Internal alkynes produce ketones, similar to those formed using H2O/H2SO4, but terminal alkynes produce aldehydes.
- As with H2O/H2SO4/HgSO4, the initial product is an enol, which then rearranges to the aldehyde or ketone.

Candela Citations
- Authored by: Martin A. Walker, Ph.D.. Provided by: SUNY Potsdam. Project: Organic Chemistry I. License: CC BY-SA: Attribution-ShareAlike